Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Rita Xu and Version 1 by Landon L Moore.
While significant strides have been made in understanding cancer biology, the enhancement in patient survival is limited, underscoring the urgency for innovative strategies. Epigenetic modifications characterized by hereditary shifts in gene expression without changes to the DNA sequence play a critical role in producing alternative gene isoforms. When these processes go awry, they influence cancer onset, growth, spread, and cancer stemness.
- epigenetics
- DCLK1
- cancer stem cells
1. Introduction
Modern curative treatments for cancer have yet to substantially improve overall survival, especially for cases with widespread metastatic disease. Even with groundbreaking advances in genomics, proteomics, and technology, wresearchers urgently need innovative platforms to revolutionize cancer therapy. The complexity of the processes and regulatory mechanisms leading to cancer is still not fully understood, especially regarding the relationship between genetics and epigenetics in the context of tumor development in specific organs. Traditionally, cancer has been viewed as the result of uncontrolled cell growth due to the activation of oncogenes and the deactivation of tumor-suppressing pathways [1]. However, new findings suggest that cancer-causing processes might start even before these genetic changes occur. Environmental factors like radiation, toxins, infections, and inflammation can cause early changes at the DNA level, even before mutations in oncogenes become evident [2,3][2][3]. These changes include alterations in DNA methylation due to reactive oxygen species (ROS) [4], inflammatory cytokines [5], and DNA repair mechanisms [6]. All these factors can influence the activity of genes that suppress or promote tumor growth (Figure 1). Moreover, epigenetic changes, which are alterations that do not change the DNA sequence but affect gene expression, are pivotal in forming cancer stem cells (CSCs). These cells are implicated in tumor diversity, resistance to treatment, recurrence, immune system evasion, and metastasis [7,8][7][8]. With the help of advanced molecular techniques, wresearchers now see an even more complex landscape of genetic and epigenetic interactions driving cancer. A particular point of interest in this landscape is genes that have isoforms, or variants, that can either promote or suppress tumor growth depending on which version is active. An example of this is the gene doublecortin-like kinase-1 (DCLK1), a well-recognized marker for CSCs [9,10,11][9][10][11] that is linked with aggressive cancer types and resistance to treatment [12,13][12][13]. DCLK1 has multiple isoforms, and recent research suggests that they might have contrasting roles in cancer and other diseases [14]. Furthermore, these isoforms may be modulated via differential DNA methylation [15].
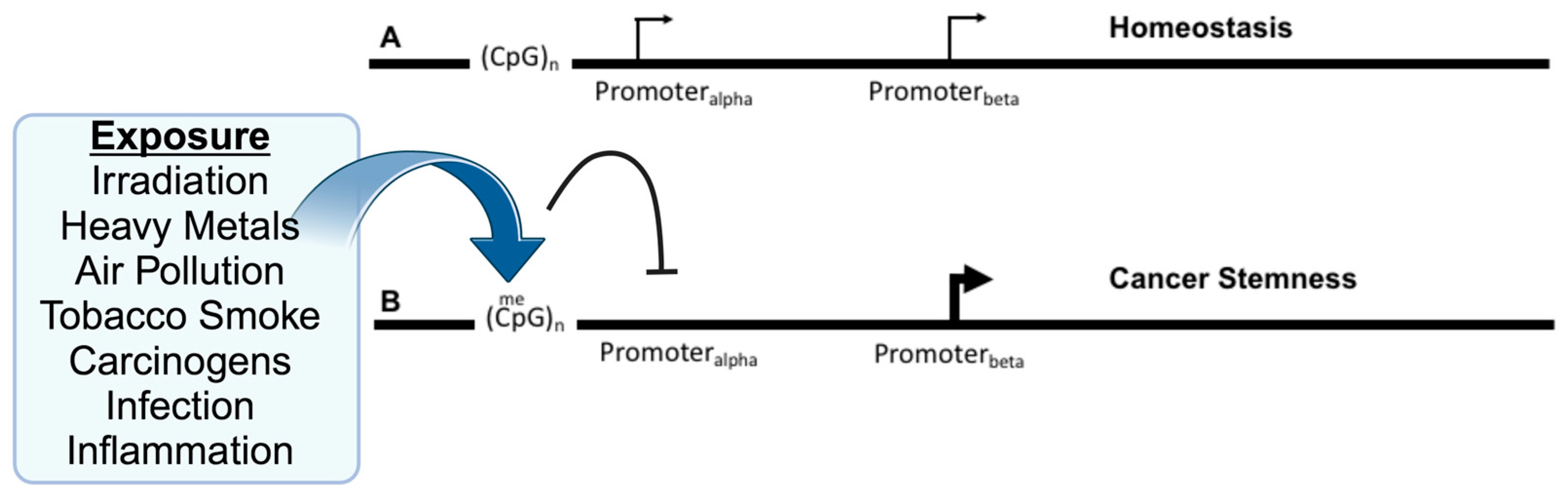
Figure 1. Exposure to environmental insults, such as irradiation or inflammation, alter DNA methylation and gene isoforms to promote cancer. (A) Several genes have been identified as having alternative promoters, Promoteralpha and Promoterbeta, with the upstream promoter located near CpG-islands that are the target of DNA methylation. Expression may be primarily from the upstream promoter, but this does not preclude expression from the downstream promoter, which may reflect the homeostatic balance of the individual isoforms and their roles. (B) Upon various environmental exposures, the DNA methylation of the upstream promoter is hypermethylated at nearby CpG sites resulting in the inhibition (arc with crossbar) of the promoter. This may directly or indirectly result in an increase in expression from the downstream promoter, which may affect tumorigenesis via the imbalance between the isoforms and their relative functions.
2. Gene Isoforms in Cancer: A Double-Edged Sword
Gene regulation becomes more intricate due to alternative promoters, leading to diverse protein isoforms that significantly sway cell behavior. This phenomenon, known as promoter choice, results in the creation of unique mRNA and protein isoforms. These isoforms can sometimes perform contrasting functions [34][16]. Cancer development relies on the balance between oncogenes, which promote cell growth, and tumor suppressors, which restrain uncontrolled cell division. Notable oncogenes include MYC, KRAS, and EGFR, while TP53, BRCA1/2, and PTEN are well-known tumor suppressors [1,55][1][17]. This balance is foundational to understanding cancer’s molecular nature and shapes the design of therapies targeting these proteins [56][18].
Tumorigenesis is multi-faceted, characterized by both genetic and epigenetic changes that lead to unregulated cell growth. One pivotal component in this complex landscape is gene isoforms, which are various forms of the same gene. Surprisingly, some genes can yield isoforms that act as both oncogenes and tumor suppressors. For instance, members of the p53 family, TP63 and TP73, produce TAp63/TAp73, which resemble tumor suppressors, and ΔNp63/73, which might have oncogenic properties [57,58][19][20]. Similarly, the RASSF1 gene presents RASSF1A as a tumor suppressor and RASSF1C with potential oncogenic roles [59][21]. The regulation of these isoforms can be influenced by epigenetic modifications, introducing another layer of intricacy in their roles in cancer [8].
One key protein, DCLK1, known to regulate CSCs, is gaining attention in cancer research (reviewed by Chhetri et al. [60][22]). It is linked to the self-renewal and tumorigenic abilities of CSCs in various cancers [61,62,63][23][24][25]. Epigenetic mechanisms, like DNA methylation and histone modifications, can regulate the expression of DCLK1 and the stem-like properties of CSCs, affecting tumorigenesis and treatment responses [64][26]. A growing body of evidence suggests that DCLK1 is a marker for CSCs and a promising target for therapies aiming to halt tumor recurrence and metastasis [65,66,67][27][28][29]. The presence of alternative promoters complicates gene regulation, resulting in a plethora of protein isoforms pivotal in cancer development. This intricate dance between oncogenes and tumor suppressor genes is central to the molecular essence of cancer and is invaluable for therapeutic development.
3. Epigenetic Regulation of Alternative Isoforms in Cancer
Gene isoforms can function as either oncogenes or tumor suppressors, based on their context and differential expression. Epigenetic regulation, notably DNA methylation and histone modifications, often governs the expression of these isoforms. Examples include RASSF1, TP73, and DCLK1 genes [68][30].
3.1. RASSF1
RASSF1 plays a key role in cell cycle regulation and has implications in various cancers. The gene yields two main isoforms, RASSF1A and RASSF1C, through alternative promoter usage [59,69][21][31]. RASSF1A, a known tumor suppressor, is often silenced in many malignancies [70][32]. Additionally, the RASSF1 promoter methylation status serves as a diagnostic and prognostic marker in breast, lung, and ovarian cancer [71,72,73][33][34][35]. On the other hand, RASSF1C is believed to have oncogenic potential [59][21]. Importantly, the two isoforms differ in a protein domain that influences their role in apoptosis and cellular proliferation. The RASSF1 5′ promoter responsible for RASSF1A contains a CpG island, which, when hypermethylated, leads to transcriptional silencing of the gene [69][31], while, in contrast, the downstream promoter that generates RASSF1C is less frequently methylated.
The fundamental difference between RASSF1A and RASSF1C is that RASSF1A contains a protein kinase C domain lacking in RASSF1C [59][21]. One consequence is that RASSF1C does not associate with death receptor complexes the same way as RASSF1A, leading to the inhibition of apoptosis and promotion of the upregulation of proliferation and invasive phenotypes [74][36]. Thus, these divergent roles are due to the absence of a single domain that is important in regulating physical interactions [75,76][37][38].
3.2. TP63/TP73
This theme of one isoform lacking a functional domain that determines its role in cancer is also seen in the TP63/TP73 gene that also has multiple gene isoforms that play a crucial role in multiple cellular functions including differentiation, proliferation, and apoptosis. Both TP63 and TP73 resemble the well-studied p53 tumor suppressor gene but exhibit structurally and functionally distinct isoforms arising from alternative promoters and extensive alternative splicing [77][39]. In both TP63 and TP73 gene isoforms, the upstream promoter (P1) drives the transcription of full-length or transactivating (TA) isoforms, which have a similar structure to p53 and encompass transcriptional activation, DNA binding, and oligomerization domains [57][19]. The TA isoforms often function as tumor suppressors, promoting cell cycle arrest and apoptosis. On the other hand, the alternative internal promoter (P2) drives the expression of N-terminally truncated (∆N) isoforms that lack the transcriptional activation domain and act in a dominant-negative manner, inhibiting the function of TA isoforms and promoting tumorigenesis [57][19]. Furthermore, CpG methylation within the P1 promoter region has been reported, resulting in transcriptional repression of the TA isoforms [78,79][40][41].
3.3. DCLK1
DCLK1 presents several isoforms due to alternative promoters and mRNA processing. Although their specific roles in cancer are yet to be fully understood, DCLK1.4 seems to be particularly crucial. Unlike TP63/TP73 where loss of the transcriptional activation domain allows for a dominant negative effect, DCLK1 is not known to form multimers; however, DCLK1 may be more like RASSF1 in that the different isoforms interact with different complexes to modulate their activity. DCLK1 is associated with CSCs [9,10,11][9][10][11] and promotes characteristics linked with aggressive cancer [12,13][12][13]. DCLK1 kinase inhibitors have shown promise in reducing tumorigenesis [80,81,82][42][43][44]. However, antibodies directed against a non-kinase extracellular domain also showed significant effects on tumor progression [83,84][45][46]. These results suggest that DCLK1 may also have alternative domains present in distinct isoforms that define its role in cancer progression.
DCLK1 was first described as a brain-specific protein with similarity to the previously identified doublecortin (DCX) gene [85,86,87,88][47][48][49][50]. Both DCLK1 and DCX have a pair of domains required for tubulin binding and microtubule assembly [89,90][51][52]. However, DCLK1 also has a domain resembling the Ca2+/calmodulin-dependent protein kinase indicating additional functionality [87,91][49][53]. The DCLK1 locus generates several isoforms using alternative promoters and alternative mRNA processing (Figure 2) [14]. Interestingly, DCLK1 can be proteolytically processed into a shorter form lacking the microtubule binding domains, indicating a possible functional difference between DCX-containing and non-DCX fragments of the protein [92,93][54][55]. Moreover, DCLK1 isoforms are differentially expressed and localized in developing mouse brains [94][56]. This study also showed that DCLK1 isoforms were generated via transcription, indicating regulation by distinct promoters. Furthermore, a study using isoform-specific antibodies found that the different isoforms tended to accumulate in different locations, suggesting that localization is important for distinctive functions [95][57]. Given that different DCLK1 promoters generate unique isoforms and that these isoforms have altered functional domains, it is likely that the DCLK1 isoforms have unique functions.
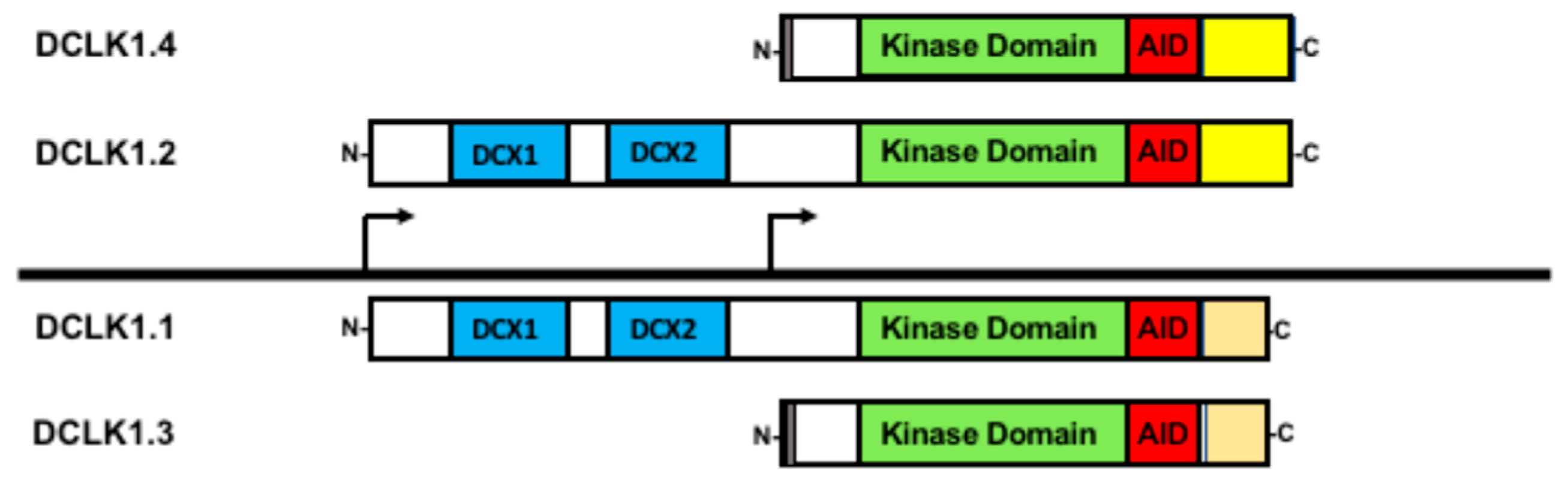
Figure 2. Doublecortin-like kinase-1 (DCLK1) isoforms generated via alternative promoter and alternative RNA splicing. All the four main isoforms contain the kinase domain (green) and its autoregulatory domain (AID) shown in red. Isoforms DCLK1.1 and DCLK1.2 (often referred as DCLK1-L) both contain the microtubule binding domains (DCX, shown in blue) yet differ in their C-terminal domains due to alternative RNA processing (yellow versus orange). From the downstream promoter, two isoforms (sometimes referenced as DCLK1-S) are generated also with distinct C-termini from alternative RNA processing, providing a further means of functional regulation. A short sequence at the N-terminus unique to the two downstream promoter products is shown in grey.
DCLK1 is transcribed from two distinct promoters [14,15][14][15]. The 5′ most promoter, termed the alpha-promoter (DCLK1alpha), produces two full-length transcripts encoding proteins that contain both the microtubule-binding doublecortin domains and the Ca2+/calmodulin-dependent protein kinase domain, often referred to as DCLK1-long (DCLK1-L). However, alternative processing, whereby a specific exon is either retained or skipped, distinguishes DCLK1-L into two isoforms that differ in their C-termini [14]; wresearchers refer to DCLK1-L with the exon retained as DCLK1.1 and the variant with the exon skipped as DCLK1.2. Because the alternative processing leads to a change in the respective open reading frame, DCLK1.2 is longer than DCLK1.1 and is different in its final C-terminal sequence, providing a way to distinguish between these two isoforms (Figure 2). Furthermore, the expression of DCLK1.1 and DCLK1.2, being driven by the same promoter, at least transcriptionally, is coordinated. Studies in colorectal, pancreatic, gastric, and lung cancers have indicated that this promoter may be epigenetically silenced through hypermethylation [13,15,96,97][13][15][58][59]. In cases of DCLK1alpha promoter silencing, DCLK1 is still observed due to a second promoter, termed the beta promoter (DCLK1beta), present downstream of the DCLK1alpha promoter and located in an intron. This DCLK1beta promoter produces transcripts that are translated into proteins lacking the microtubule-binding domain and are termed DCLK1-short (DCLK1-S) [14,98][14][60]. The same potential for alternative splicing as in DCLK1-L occurs in DCLK1-S [98][60], and wresearchers refer to these two isoforms as DCLK1.3 (retained exon) and DCLK1.4 (skipped exon). Furthermore, DCLK1.4 was independently isolated in rats as the candidate plasticity gene (CPG16) supporting the conclusion that the DCLK1beta promoter and its products are biologically relevant [91][53]. The DCLK1beta promoter lacks prominent CpG islands with which to be silenced and appears that it may be regulated by NF-kBp65 [15] and FOXD3 [99][61]. Furthermore, the DCLK1beta promoter is also regulated by lymphoid enhancer-binding factor (LEF1) [100][62]. However, it remains unclear whether promoter usage regulates the alternative splicing or that the alternative splicing itself is subject to regulation to generate these DCLK1 isoforms.
Numerous studies have implicated the high expression of DCLK1 with poor prognosis and that the inhibition of DCLK1 via a variety of methods suggests that targeting DCLK1 is a viable strategy for cancer therapy [60][22]. Recent work has focused on the role of different isoforms of DCLK1 in cancer progression. Unfortunately, most of these studies did not distinguish between the various DCLK1 isoforms (see Kalantari et al. [101][63]). What is identifiable is that DCLK1 modulates several pathways important in cancer progression, such as the epithelial-to-mesenchymal transition (EMT), cancer stemness, inflammation, and metastasis. Furthermore, the overexpression of DCLK1.2 was seen to increase cancer aggressiveness in PDAC and that targeting DCLK1.2’s unique C-terminal domain could inhibit tumorigenesis [102][64]. It has also been linked to tumor immunosuppression via M2-macrophage polarization [103][65]. An important caveat to these types of overexpression studies with kinases is that these studies represent unfettered kinase activity that may obscure the subtle aspects of kinase regulation [104][66].
Knockdown studies and DCLK1 kinase inhibitor studies have shown improvements in reducing tumorigenesis and modulating the tumor microenvironment [105][67]. However, most of these studies are broad-spectrum, affecting all isoforms of DCLK1 as they all retain the kinase domain, raising the question of whether dysregulated or overactive DCLK1 kinase is the root cause. Interestingly, treatments targeting the C-terminal domain of DCLK1.2 and DCLK1.4 show significant effects on cancer progression, suggesting that the C-terminal domain is important [84][46]. It is not uncommon for kinases to be regulated by a C-terminal domain and, indeed, a C-terminal autoinhibitory domain (AID) common to all isoforms was identified that regulates DCLK1 autophosphorylation [104,106][66][68]. However, an antibody CBT-15 against the unique C-terminal domain of DCLK1.2/DCLK1.4 outside of the AID domain in the intrinsically disordered domain could inhibit tumor progression, indicating the existence of other regulatory domains [83,84][45][46].
Recent work has focused on the oncogenic role of the DCLK1-S isoform [99,107][61][69]. DCLK1.4 is detected in CSCs, suggesting a role in their formation and/or maintenance [95,100][57][62]. Furthermore, the inhibition of DCLK1.4 transcription via the DCLK1beta promoter through blocking LEF-1 diminishes cancer stemness [100][62]. DCLK1.4 also induces the EMT, a key property related to the increased aggressiveness of cancer and immune suppression [108,109][70][71]. DCLK1 regulates inflammation in both human and murine colitis with DCLK1.2 and DCLK1.4 showing differential regulation [68][30]. Additionally, these changes were correlated with a decrease in FoxD3, an inhibitor of the DCLK1beta promoter, suggesting that DCLK1.4 upregulation acts to promote inflammation, a known driver of tumorigenesis, and that the balance between DCLK1.2 and DCLK1.4 is important. In colorectal cancer, DCLK1.4 was shown to promote cancer stemness and aggressiveness, consistent with its overexpression relative to DCLK1.2 [9,15][9][15]. This was dependent on the DCLK1 phosphorylation of XRCC5 and the co-option of the inflammatory tumor microenvironment (TME) [9]. Given the results that DCLK1.4 isoform overexpression is highly oncogenic, while DCLK1.2, through its silencing, fits the definition of a tumor suppressor, the restoration of DCLK1.2/DCLK1.4 balance most importantly appears to be vital for blocking cancer progression.
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674.
- Sabit, H.; Cevik, E.; Tombuloglu, H. Colorectal cancer: The epigenetic role of microbiome. World J. Clin. Cases 2019, 7, 3683–3697.
- Sun, D.; Chen, Y.; Fang, J.Y. Influence of the microbiota on epigenetics in colorectal cancer. Natl. Sci. Rev. 2019, 6, 1138–1148.
- Wu, Q.; Ni, X. ROS-mediated DNA methylation pattern alterations in carcinogenesis. Curr. Drug Targets 2015, 16, 13–19.
- Das, D.; Karthik, N.; Taneja, R. Crosstalk Between Inflammatory Signaling and Methylation in Cancer. Front. Cell Dev. Biol. 2021, 9, 756458.
- O’Hagan, H.M.; Mohammad, H.P.; Baylin, S.B. Double strand breaks can initiate gene silencing and SIRT1-dependent onset of DNA methylation in an exogenous promoter CpG island. PLoS Genet. 2008, 4, e1000155.
- Baylin, S.B.; Jones, P.A. A decade of exploring the cancer epigenome—Biological and translational implications. Nat. Rev. Cancer 2011, 11, 726–734.
- Easwaran, H.; Tsai, H.C.; Baylin, S.B. Cancer epigenetics: Tumor heterogeneity, plasticity of stem-like states, and drug resistance. Mol. Cell 2014, 54, 716–727.
- Kim, J.H.; Park, S.Y.; Jeon, S.E.; Choi, J.H.; Lee, C.J.; Jang, T.Y.; Yun, H.J.; Lee, Y.; Kim, P.; Cho, S.H.; et al. DCLK1 promotes colorectal cancer stemness and aggressiveness via the XRCC5/COX2 axis. Theranostics 2022, 12, 5258–5271.
- Kalantari, E.; Ghods, R.; Saeednejad Zanjani, L.; Rahimi, M.; Eini, L.; Razmi, M.; Asadi-Lari, M.; Madjd, Z. Cytoplasmic expression of DCLK1-S, a novel DCLK1 isoform, is associated with tumor aggressiveness and worse disease-specific survival in colorectal cancer. Cancer Biomark. 2022, 33, 277–289.
- Wang, L.; Zhao, L.; Lin, Z.; Yu, D.; Jin, M.; Zhou, P.; Ren, J.; Cheng, J.; Yang, K.; Wu, G.; et al. Targeting DCLK1 overcomes 5-fluorouracil resistance in colorectal cancer through inhibiting CCAR1/beta-catenin pathway-mediated cancer stemness. Clin. Transl. Med. 2022, 12, e743.
- Chandrakesan, P.; Weygant, N.; May, R.; Qu, D.; Chinthalapally, H.R.; Sureban, S.M.; Ali, N.; Lightfoot, S.A.; Umar, S.; Houchen, C.W. DCLK1 facilitates intestinal tumor growth via enhancing pluripotency and epithelial mesenchymal transition. Oncotarget 2014, 5, 9269–9280.
- Vedeld, H.M.; Skotheim, R.I.; Lothe, R.A.; Lind, G.E. The recently suggested intestinal cancer stem cell marker DCLK1 is an epigenetic biomarker for colorectal cancer. Epigenetics 2014, 9, 346–350.
- Burgess, H.A.; Reiner, O. Alternative splice variants of doublecortin-like kinase are differentially expressed and have different kinase activities. J. Biol. Chem. 2002, 277, 17696–17705.
- O’Connell, M.R.; Sarkar, S.; Luthra, G.K.; Okugawa, Y.; Toiyama, Y.; Gajjar, A.H.; Qiu, S.; Goel, A.; Singh, P. Epigenetic changes and alternate promoter usage by human colon cancers for expressing DCLK1-isoforms: Clinical Implications. Sci. Rep. 2015, 5, 14983.
- Davuluri, R.V.; Suzuki, Y.; Sugano, S.; Plass, C.; Huang, T.H. The functional consequences of alternative promoter use in mammalian genomes. Trends Genet. 2008, 24, 167–177.
- Knudson, A.G. Two genetic hits (more or less) to cancer. Nat. Rev. Cancer 2001, 1, 157–162.
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A., Jr.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558.
- Rodriguez Calleja, L.; Lavaud, M.; Tesfaye, R.; Brounais-Le-Royer, B.; Baud’huin, M.; Georges, S.; Lamoureux, F.; Verrecchia, F.; Ory, B. The p53 Family Members p63 and p73 Roles in the Metastatic Dissemination: Interactions with microRNAs and TGFbeta Pathway. Cancers 2022, 14, 5948.
- Xu, Y.; Yang, X.; Xiong, Q.; Han, J.; Zhu, Q. The dual role of p63 in cancer. Front. Oncol. 2023, 13, 1116061.
- Donninger, H.; Vos, M.D.; Clark, G.J. The RASSF1A tumor suppressor. J. Cell Sci. 2007, 120, 3163–3172.
- Chhetri, D.; Vengadassalapathy, S.; Venkadassalapathy, S.; Balachandran, V.; Umapathy, V.R.; Veeraraghavan, V.P.; Jayaraman, S.; Patil, S.; Iyaswamy, A.; Palaniyandi, K.; et al. Pleiotropic effects of DCLK1 in cancer and cancer stem cells. Front. Mol. Biosci. 2022, 9, 965730.
- Nakanishi, Y.; Seno, H.; Fukuoka, A.; Ueo, T.; Yamaga, Y.; Maruno, T.; Nakanishi, N.; Kanda, K.; Komekado, H.; Kawada, M.; et al. Dclk1 distinguishes between tumor and normal stem cells in the intestine. Nat. Genet. 2013, 45, 98–103.
- Bailey, J.M.; Alsina, J.; Rasheed, Z.A.; McAllister, F.M.; Fu, Y.Y.; Plentz, R.; Zhang, H.; Pasricha, P.J.; Bardeesy, N.; Matsui, W.; et al. DCLK1 marks a morphologically distinct subpopulation of cells with stem cell properties in preinvasive pancreatic cancer. Gastroenterology 2014, 146, 245–256.
- Westphalen, C.B.; Asfaha, S.; Hayakawa, Y.; Takemoto, Y.; Lukin, D.J.; Nuber, A.H.; Brandtner, A.; Setlik, W.; Remotti, H.; Muley, A.; et al. Long-lived intestinal tuft cells serve as colon cancer-initiating cells. J. Clin. Investig. 2014, 124, 1283–1295.
- Sureban, S.M.; May, R.; Lightfoot, S.A.; Hoskins, A.B.; Lerner, M.; Brackett, D.J.; Postier, R.G.; Ramanujam, R.; Mohammed, A.; Rao, C.V.; et al. DCAMKL-1 regulates epithelial-mesenchymal transition in human pancreatic cells through a miR-200a-dependent mechanism. Cancer Res. 2011, 71, 2328–2338.
- Standing, D.; Arnold, L.; Dandawate, P.; Ottemann, B.; Snyder, V.; Ponnurangam, S.; Sayed, A.; Subramaniam, D.; Srinivasan, P.; Choudhury, S.; et al. Doublecortin-like kinase 1 is a therapeutic target in squamous cell carcinoma. Mol. Carcinog. 2023, 62, 145–159.
- Vijai, M.; Baba, M.; Ramalingam, S.; Thiyagaraj, A. DCLK1 and its interaction partners: An effective therapeutic target for colorectal cancer. Oncol. Lett. 2021, 22, 850.
- Liu, H.; Yan, R.; Xiao, Z.; Huang, X.; Yao, J.; Liu, J.; An, G.; Ge, Y. Targeting DCLK1 attenuates tumor stemness and evokes antitumor immunity in triple-negative breast cancer by inhibiting IL-6/STAT3 signaling. Breast Cancer Res. 2023, 25, 43.
- Roy, B.C.; Ahmed, I.; Stubbs, J.; Zhang, J.; Attard, T.; Septer, S.; Welch, D.; Anant, S.; Sampath, V.; Umar, S. DCLK1 isoforms and aberrant Notch signaling in the regulation of human and murine colitis. Cell Death Discov. 2021, 7, 169.
- Dammann, R.; Li, C.; Yoon, J.H.; Chin, P.L.; Bates, S.; Pfeifer, G.P. Epigenetic inactivation of a RAS association domain family protein from the lung tumour suppressor locus 3p21.3. Nat. Genet. 2000, 25, 315–319.
- Pfeifer, G.P.; Yoon, J.H.; Liu, L.; Tommasi, S.; Wilczynski, S.P.; Dammann, R. Methylation of the RASSF1A gene in human cancers. Biol. Chem. 2002, 383, 907–914.
- da Costa Prando, E.; Cavalli, L.R.; Rainho, C.A. Evidence of epigenetic regulation of the tumor suppressor gene cluster flanking RASSF1 in breast cancer cell lines. Epigenetics 2011, 6, 1413–1424.
- Zhang, T.; Li, Y.; Zhang, H.; Wang, X.; Liu, X.; Li, L. The Role of RASSF1 Methylation in Lung Carcinoma. Adv. Exp. Med. Biol. 2020, 1255, 99–108.
- Rattanapan, Y.; Korkiatsakul, V.; Kongruang, A.; Chareonsirisuthigul, T.; Rerkamnuaychoke, B.; Wongkularb, A.; Wilailak, S. EGFL7 and RASSF1 promoter hypermethylation in epithelial ovarian cancer. Cancer Genet. 2018, 224–225, 37–40.
- Dubois, F.; Bergot, E.; Levallet, G. Cancer and RASSF1A/RASSF1C, the Two Faces of Janus. Trends Cancer 2019, 5, 662–665.
- Reeves, M.E.; Baldwin, S.W.; Baldwin, M.L.; Chen, S.T.; Moretz, J.M.; Aragon, R.J.; Li, X.; Strong, D.D.; Mohan, S.; Amaar, Y.G. Ras-association domain family 1C protein promotes breast cancer cell migration and attenuates apoptosis. BMC Cancer 2010, 10, 562.
- Ortiz-Vega, S.; Khokhlatchev, A.; Nedwidek, M.; Zhang, X.F.; Dammann, R.; Pfeifer, G.P.; Avruch, J. The putative tumor suppressor RASSF1A homodimerizes and heterodimerizes with the Ras-GTP binding protein Nore1. Oncogene 2002, 21, 1381–1390.
- Osterburg, C.; Dotsch, V. Structural diversity of p63 and p73 isoforms. Cell Death Differ. 2022, 29, 921–937.
- Lai, J.; Nie, W.; Zhang, W.; Wang, Y.; Xie, R.; Wang, Y.; Gu, J.; Xu, J.; Song, W.; Yang, F.; et al. Transcriptional regulation of the p73 gene by Nrf-2 and promoter CpG methylation in human breast cancer. Oncotarget 2014, 5, 6909–6922.
- Pokorna, Z.; Hrabal, V.; Tichy, V.; Vojtesek, B.; Coates, P.J. DNA Demethylation Switches Oncogenic ΔNp63 to Tumor Suppressive TAp63 in Squamous Cell Carcinoma. Front. Oncol. 2022, 12, 924354.
- Ding, L.; Yang, Y.; Ge, Y.; Lu, Q.; Yan, Z.; Chen, X.; Du, J.; Hafizi, S.; Xu, X.; Yao, J.; et al. Inhibition of DCLK1 with DCLK1-IN-1 Suppresses Renal Cell Carcinoma Invasion and Stemness and Promotes Cytotoxic T-Cell-Mediated Anti-Tumor Immunity. Cancers 2021, 13, 5729.
- Patel, O.; Roy, M.J.; Kropp, A.; Hardy, J.M.; Dai, W.; Lucet, I.S. Structural basis for small molecule targeting of Doublecortin Like Kinase 1 with DCLK1-IN-1. Commun. Biol. 2021, 4, 1105.
- Ferguson, F.M.; Liu, Y.; Harshbarger, W.; Huang, L.; Wang, J.; Deng, X.; Capuzzi, S.J.; Muratov, E.N.; Tropsha, A.; Muthuswamy, S.; et al. Synthesis and Structure-Activity Relationships of DCLK1 Kinase Inhibitors Based on a 5,11-Dihydro-6H-benzopyrimidodiazepin-6-one Scaffold. J. Med. Chem. 2020, 63, 7817–7826.
- Sureban, S.M.; Berahovich, R.; Zhou, H.; Xu, S.; Wu, L.; Ding, K.; May, R.; Qu, D.; Bannerman-Menson, E.; Golubovskaya, V.; et al. DCLK1 Monoclonal Antibody-Based CAR-T Cells as a Novel Treatment Strategy against Human Colorectal Cancers. Cancers 2019, 12, 54.
- Ge, Y.; Weygant, N.; Qu, D.; May, R.; Berry, W.L.; Yao, J.; Chandrakesan, P.; Zheng, W.; Zhao, L.; Zhao, K.L.; et al. Alternative splice variants of DCLK1 mark cancer stem cells, promote self-renewal and drug-resistance, and can be targeted to inhibit tumorigenesis in kidney cancer. Int. J. Cancer 2018, 143, 1162–1175.
- Omori, Y.; Suzuki, M.; Ozaki, K.; Harada, Y.; Nakamura, Y.; Takahashi, E.; Fujiwara, T. Expression and chromosomal localization of KIAA0369, a putative kinase structurally related to Doublecortin. J. Hum. Genet. 1998, 43, 169–177.
- Burgess, H.A.; Martinez, S.; Reiner, O. KIAA0369, doublecortin-like kinase, is expressed during brain development. J. Neurosci. Res. 1999, 58, 567–575.
- Matsumoto, N.; Pilz, D.T.; Ledbetter, D.H. Genomic structure, chromosomal mapping, and expression pattern of human DCAMKL1 (KIAA0369), a homologue of DCX (XLIS). Genomics 1999, 56, 179–183.
- Sossey-Alaoui, K.; Srivastava, A.K. DCAMKL1, a brain-specific transmembrane protein on 13q12.3 that is similar to doublecortin (DCX). Genomics 1999, 56, 121–126.
- Moores, C.A.; Perderiset, M.; Francis, F.; Chelly, J.; Houdusse, A.; Milligan, R.A. Mechanism of microtubule stabilization by doublecortin. Mol. Cell 2004, 14, 833–839.
- Lin, P.T.; Gleeson, J.G.; Corbo, J.C.; Flanagan, L.; Walsh, C.A. DCAMKL1 encodes a protein kinase with homology to doublecortin that regulates microtubule polymerization. J. Neurosci. 2000, 20, 9152–9161.
- Silverman, M.A.; Benard, O.; Jaaro, H.; Rattner, A.; Citri, Y.; Seger, R. CPG16, a novel protein serine/threonine kinase downstream of cAMP-dependent protein kinase. J. Biol. Chem. 1999, 274, 2631–2636.
- Burgess, H.A.; Reiner, O. Cleavage of doublecortin-like kinase by calpain releases an active kinase fragment from a microtubule anchorage domain. J. Biol. Chem. 2001, 276, 36397–36403.
- Hardt, R.; Dehghani, A.; Schoor, C.; Gödderz, M.; Cengiz Winter, N.; Ahmadi, S.; Sharma, R.; Schork, K.; Eisenacher, M.; Gieselmann, V.; et al. Proteomic investigation of neural stem cell to oligodendrocyte precursor cell differentiation reveals phosphorylation-dependent Dclk1 processing. Cell. Mol. Life Sci. 2023, 80, 260.
- Bergoglio, E.; Suzuki, I.K.; Togashi, K.; Tsuji, M.; Takeuchi, S.; Koizumi, H.; Emoto, K. Spatial and temporal diversity of DCLK1 isoforms in developing mouse brain. Neurosci. Res. 2021, 170, 154–165.
- Sarkar, S.; Popov, V.L.; O’Connell, M.R.; Stevenson, H.L.; Lee, B.S.; Obeid, R.A.; Luthra, G.K.; Singh, P. A novel antibody against cancer stem cell biomarker, DCLK1-S, is potentially useful for assessing colon cancer risk after screening colonoscopy. Lab. Investig. 2017, 97, 1245–1261.
- Vedeld, H.M.; Andresen, K.; Eilertsen, I.A.; Nesbakken, A.; Seruca, R.; Gladhaug, I.P.; Thiis-Evensen, E.; Rognum, T.O.; Boberg, K.M.; Lind, G.E. The novel colorectal cancer biomarkers CDO1, ZSCAN18 and ZNF331 are frequently methylated across gastrointestinal cancers. Int. J. Cancer 2015, 136, 844–853.
- Powrozek, T.; Krawczyk, P.; Nicos, M.; Kuznar-Kaminska, B.; Batura-Gabryel, H.; Milanowski, J. Methylation of the DCLK1 promoter region in circulating free DNA and its prognostic value in lung cancer patients. Clin. Transl. Oncol. 2016, 18, 398–404.
- Engels, B.M.; Schouten, T.G.; van Dullemen, J.; Gosens, I.; Vreugdenhil, E. Functional differences between two DCLK splice variants. Brain Res. Mol. Brain Res. 2004, 120, 103–114.
- Sarkar, S.; O’Connell, M.R.; Okugawa, Y.; Lee, B.S.; Toiyama, Y.; Kusunoki, M.; Daboval, R.D.; Goel, A.; Singh, P. FOXD3 Regulates CSC Marker, DCLK1-S, and Invasive Potential: Prognostic Implications in Colon Cancer. Mol. Cancer Res. 2017, 15, 1678–1691.
- Park, S.Y.; Kim, J.Y.; Choi, J.H.; Kim, J.H.; Lee, C.J.; Singh, P.; Sarkar, S.; Baek, J.H.; Nam, J.S. Inhibition of LEF1-Mediated DCLK1 by Niclosamide Attenuates Colorectal Cancer Stemness. Clin. Cancer Res. 2019, 25, 1415–1429.
- Kalantari, E.; Razmi, M.; Tajik, F.; Asadi-Lari, M.; Ghods, R.; Madjd, Z. Oncogenic functions and clinical significances of DCLK1 isoforms in colorectal cancer: A systematic review and meta-analysis. Cancer Cell Int. 2022, 22, 217.
- Qu, D.; Weygant, N.; Yao, J.; Chandrakesan, P.; Berry, W.L.; May, R.; Pitts, K.; Husain, S.; Lightfoot, S.; Li, M.; et al. Overexpression of DCLK1-AL Increases Tumor Cell Invasion, Drug Resistance, and KRAS Activation and Can Be Targeted to Inhibit Tumorigenesis in Pancreatic Cancer. J. Oncol. 2019, 2019, 6402925.
- Chandrakesan, P.; Panneerselvam, J.; May, R.; Weygant, N.; Qu, D.; Berry, W.R.; Pitts, K.; Stanger, B.Z.; Rao, C.V.; Bronze, M.S.; et al. DCLK1-Isoform2 Alternative Splice Variant Promotes Pancreatic Tumor Immunosuppressive M2-Macrophage Polarization. Mol. Cancer Ther. 2020, 19, 1539–1549.
- Cheng, L.; Yang, Z.; Guo, W.; Wu, C.; Liang, S.; Tong, A.; Cao, Z.; Thorne, R.F.; Yang, S.Y.; Yu, Y.; et al. DCLK1 autoinhibition and activation in tumorigenesis. Innovation 2022, 3, 100191.
- Cao, Z.; Weygant, N.; Chandrakesan, P.; Houchen, C.W.; Peng, J.; Qu, D. Tuft and Cancer Stem Cell Marker DCLK1: A New Target to Enhance Anti-Tumor Immunity in the Tumor Microenvironment. Cancers 2020, 12, 3801.
- Agulto, R.L.; Rogers, M.M.; Tan, T.C.; Ramkumar, A.; Downing, A.M.; Bodin, H.; Castro, J.; Nowakowski, D.W.; Ori-McKenney, K.M. Autoregulatory control of microtubule binding in doublecortin-like kinase 1. eLife 2021, 10, e60126.
- Takiyama, A.; Tanaka, T.; Kazama, S.; Nagata, H.; Kawai, K.; Hata, K.; Otani, K.; Nishikawa, T.; Sasaki, K.; Kaneko, M.; et al. DCLK1 Expression in Colorectal Polyps Increases with the Severity of Dysplasia. In Vivo 2018, 32, 365–371.
- Ge, Y.; Fan, X.; Huang, X.; Weygant, N.; Xiao, Z.; Yan, R.; Liu, H.; Liu, J.; An, G.; Yao, J. DCLK1-Short Splice Variant Promotes Esophageal Squamous Cell Carcinoma Progression via the MAPK/ERK/MMP2 Pathway. Mol. Cancer Res. 2021, 19, 1980–1991.
- Ge, Y.; Liu, H.; Zhang, Y.; Liu, J.; Yan, R.; Xiao, Z.; Fan, X.; Huang, X.; An, G. Inhibition of DCLK1 kinase reverses epithelial-mesenchymal transition and restores T-cell activity in pancreatic ductal adenocarcinoma. Transl. Oncol. 2022, 17, 101317.
More