Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Jamie L Archambault and Version 2 by Fanny Huang.
Serotonin (5-HT) is a bioamine that has been implicated in the pathogenesis of pulmonary hypertension (PH). The lung serves as an important site of 5-HT synthesis, uptake, and metabolism with signaling primarily regulated by tryptophan hydroxylase (TPH), the 5-HT transporter (SERT), and numerous unique 5-HT receptors. Accumulating evidence from both clinical and preclinical studies has suggested that the 5-HT signaling pathway may play an important role in neonatal cardiopulmonary transition and the development of PH in newborns. The expression of TPH, SERT, and the 5-HT receptors is developmentally regulated, with alterations resulting in pulmonary vasoconstriction and pulmonary vascular remodeling.
- serotonin
- pulmonary hypertension
- neonate
1. Introduction
Serotonin (5-HT) is a bioamine most appreciated for its role as a neurotransmitter and its effect on mood and behavior. However, the majority of 5-HT is synthesized in the periphery where 5-HT modulates numerous other biologic processes including respiratory, cardiovascular, and gastrointestinal functions [1][2][3][1,2,3]. Over the past several decades, 5-HT has emerged as a potent pulmonary vasoconstrictor and angiogenic agent that plays an important role in the development of pulmonary hypertension (PH).
5-HT is synthesized from L-tryptophan through the activity of tryptophan hydroxylase (TPH), the rate-limiting enzyme in 5-HT synthesis. TPH has two distinct isoforms. TPH1 is primarily present in the enterochromaffin cells of the small intestine and is responsible for the production of peripheral 5-HT, whereas TPH2 is present in the central and enteric nervous systems [4][5][4,5]. TPH converts L-tryptophan to 5-hydroxytroptophan (5-HTP). 5-HTP is then converted by amino acid decarboxylase (AAAD) to 5-HT (Figure 1). On first pass, 30–80% of 5-HT synthesized in the intestine is metabolized by the liver and 90% of the remainder is metabolized by the lungs. The remaining 10% is primarily stored in platelets which take up 5-HT via the 5-HT transporter (SERT) (Figure 2). Consequently, only a small amount of 5-HT freely circulates and plasma levels are extremely low [6].
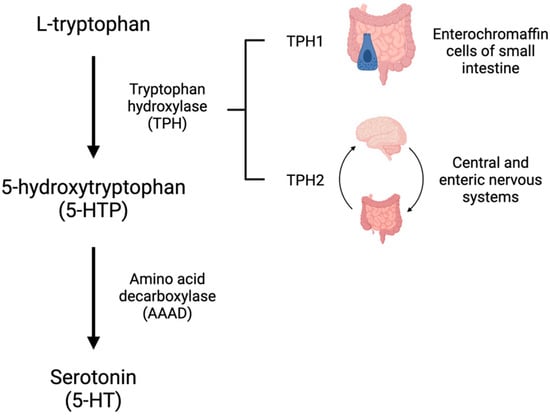
Figure 1. Serotonin (5-HT) is synthesized from the amino acid tryptophan. L-tryptophan is converted to 5-hydroxytryptophan (5-HTP) by tryptophan hydroxylase (TPH). TPH1 exists in the enterochromaffin cells of the small intestine and TPH2 is primarily found in the central and enteric nervous systems. 5-HTP is converted to 5-HT by amino acid decarboxylase (AAAD).
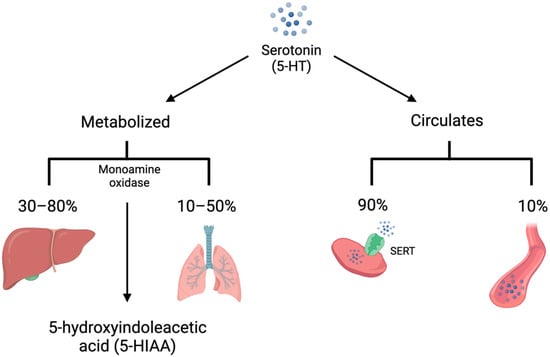
Figure 2. Serotonin (5-HT) is either metabolized by monoamine oxidase to 5-hydroxyindoleacetic acid (5-HIAA) or circulates in the bloodstream. The majority of 5-HT is metabolized on first pass by the liver, with much of the remainder metabolized by the lungs. In addition, 90% of unmetabolized 5-HT is taken up into platelets via the 5-HT transporter (SERT). Only 10% of circulating 5-HT is free in the plasma.
The diverse biologic functions of 5-HT are mediated by fifteen unique receptors divided into seven families (5-HT 1–7) based on molecular structure, signal transduction properties, and pharmacological properties [7][8][7,8]. The 5-HT 1, 2, and 4–7 receptors couple with G-proteins, while the 5-HT 3 receptors are 5-HT-gated ion channels. The altered expression and activity of both SERT and 5-HT receptors have been implicated in the pathophysiology of a wide spectrum of conditions in the fetus, newborns, and adults.
2. Serotonin in Neonatal Pulmonary Circulation
2.1. Neonatal Cardiopulmonary Transition
The definition and classification of PH are the same for children as they are for adults. While there are indeed differences in the etiology, presentation, and outcomes of pediatric PH compared to adult PH, the histopathology and mechanisms for targeted therapies remain similar [9][87]. However, it is unclear if this same framework should also be applied to infants. In utero, PVR is significantly greater than systemic vascular resistance, allowing for the preferential shunting of oxygenated placental blood to the developing fetus. At birth, ventilation of the newborn lung causes a dramatic decrease in PVR and pulmonary blood flow increases by 8–10 fold to accommodate the postnatal dependence on alveolar gas exchange [10][11][88,89]. Pulmonary artery pressures continue to decrease for 2–3 months after birth, a process that takes longer in infants born at higher altitudes. Failure to complete this physiologic transition after birth results in a persistent pulmonary hypertension of the newborn (PPHN), a disease process characterized by sustained elevation in PVR and intracardiac shunting, leading to severe hypoxemia. While the exact incidence and prevalence of PH in the pediatric population are unknown, current registries have begun to examine the etiology associated with pediatric PH. In addition to PPHN, the majority of the remaining cases of PH are caused by idiopathic PAH, PH due to congenital heart disease, or PH due to chronic lung disease of prematurity [12][13][14][90,91,92].
2.2. Selective Serotonin Reuptake Inhibitors and Persistent Pulmonary Hypertension of the Newborn
In the early 2000s, several retrospective case-control studies and meta-analyses investigated the association between the maternal use of selective serotonin reuptake inhibitors (SSRI) and the incidence of PPHN. Based on the concerning findings reported by these epidemiologic studies, the United States Food and Drug Administration issued an advisory statement warning against the potential consequences of SSRI use during pregnancy.
SSRIs are a class of medications that block the neuronal reuptake and reabsorption of 5-HT. Approximately 8–10% of pregnant women are prescribed selective SSRIs to treat anxiety and/or depression [15][16][17][93,94,95]. SSRIs bind to SERT expressed by placental syncytiotrophoblasts or cross the placenta and bind to SERT expressed by fetal platelets and organ tissues, resulting in increased local concentrations of 5-HT [18][19][20][96,97,98]. The lung, in addition to the brain, serves as a primary uptake site for SSRIs and may function as a reservoir for antidepressants with a high affinity to SERT [21][99]. SSRIs are also known to inhibit nitric oxide synthase (NOS) and may impact the concentration of nitric oxide, a potent vasodilator that plays a critical role in the regulation of vascular tone in utero and during postnatal transition [22][23][100,101]. Maternal use of SSRIs may compromise uterine/placental vascular perfusion and impact fetal growth. In humans, maternal SSRI use is associated with changes in fetal heart rate and cerebral blood flow, consistent with reductions in placental perfusion and resultant fetal hypoxia [24][102].
The early epidemiological studies found a six-fold increased risk of PPHN in neonates when exposed to maternal SSRI in pregnancy, specifically during the second and third trimesters [25][26][27][28][29][30][31][32][62,103,104,105,106,107,108,109]. However, further examination of this association revealed conflicting data, with an absolute risk difference of 0.6–2 per 1000 live births [17][33][34][35][36][37][95,110,111,112,113,114]. Clinical practice has shown the potential effects of SSRI exposure on respiratory distress are short lived, with no reported deaths. Thus, current obstetric and psychiatric/psychologic practice advises against cessation of SSRI use before or during pregnancy, acknowledging the significant maternal and infant morbidity associated with untreated perinatal depression and anxiety [38][115].
Despite conflicting evidence regarding the clinical significance of SSRI exposure and PPHN, there is a clear impact of maternal SSRI use on fetomaternal circulation [39][40][41][116,117,118]. Large animal models have shown that acute infusion of the SSRI fluoxetine into pregnant sheep leads to a transient decrease in uterine artery blood flow, fetal PaO2, and oxygen saturation [41][118]. Similarly, the offspring of rats treated with fluoxetine in late gestation displayed lower postnatal arterial oxygen saturation and PH characterized by increased pulmonary artery medial wall thickness and increased right ventricular mass [42][119].
Researchers' group utilized the chronically prepared fetal sheep model to study the in vivo hemodynamic effects of SSRIs in the developing lung. Researchers found that brief infusions of the SSRIs sertraline and fluoxetine directly to the fetal ovine lung caused potent and sustained elevation in pulmonary vascular resistance, which could be reversed by the 5-HT 2A receptor antagonist ketanserin [43][44][82,120]. Acetylcholine-induced vasodilation persisted after sertraline treatment, suggesting sertraline does not directly impair NOS activity. A similar study in mice revealed sertraline and fluoxetine contribute to in utero constriction of the ductus arteriosus, a well-known mechanism for elevated prenatal pulmonary pressures and PPHN [45][121]. Based on these mechanisms, it is plausible to conclude the accumulation of lung 5-HT from SSRI exposure could contribute to increased vasoconstriction, smooth muscle cell proliferation, and elevated pulmonary vascular resistance in exposed infants leading to the clinical syndrome known as PPHN.
2.3. Serotonin in Neonatal Pulmonary Hypertension
Several decades after the original 5-HT hypothesis was proposed in the literature, a pediatric clinical study quantified 5-HT PNECs in infants who died due to respiratory distress syndrome (RDS) and bronchopulmonary dysplasia (BPD) [46][122]. Interestingly, the number of 5-HT immunoreactive PNECs was decreased in preterm infants who died from RDS, but significantly increased in those who died from BPD. These results were most pronounced at 2 months of age, when infants with BPD had a 34-fold increase in 5-HT immunoreactive PNECs. The significance of the 5-HT pathway in the process of pulmonary vascular remodeling at birth is further supported by evidence that SERT expression is decreased in infants with alveolar capillary dysplasia with misalignment of pulmonary veins (ACD/MPV) [47][81]. ACD/MPV is a uniformly lethal condition characterized by severe PPHN. The pulmonary microvasculature of infants with ACD/MPV is markedly depleted of SERT expression, suggesting alterations in 5-HT signaling may contribute to abnormal vascular development and PH. 5-HT metabolism is also increased in pediatric patients with PH due to congenital heart disease [48][123]. In this population, there is no appreciable difference in the concentration of plasma total and free 5-HT. However, these patients have increased urinary excretion of 5-HIAA, which may be due to increased 5-HT metabolism.
Numerous preclinical studies have demonstrated an important association between 5-HT and PH in both small and large animal models of neonatal PH. 5-HT exacerbates pulmonary artery constriction in a hyperoxic rat model of BPD-induced PH, and pulmonary TPH1 is increased in both murine and ovine models of PH [44][49][50][120,124,125]. In an experimental model of congenital diaphragmatic hernia, a developmental lung disease associated with PPHN and high rates of neonatal morbidity and mortality, the expression of SERT and the 5-HT 2A receptor are upregulated in the pulmonary vasculature, suggesting 5-HT may mediate pulmonary vascular remodeling seen in this disease [51][126].
Researchers' group studied the hemodynamic effects of 5-HT and 5-HT receptor antagonists in the late-gestation ovine fetus for over a decade [43][82]. Researchers found that a brief infusion of 5-HT into the pulmonary vasculature increases PVR in a dose-dependent manner. Pharmacologic inhibition of the 5-HT 2A receptor with direct pulmonary infusions of the 5-HT 2A receptor antagonist ketanserin results in pulmonary vasodilation, suggesting endogenous 5-HT contributes to the maintenance of normal pulmonary vascular tone in the ovine fetus via activation of the 5-HT 2A receptor. Ketanserin also prevents exogenous 5-HT-induced pulmonary vasoconstriction. However, pharmacologic inhibition of the 5-HT 1B and 2B receptors does not have this effect. Researchers concluded from this work that the pulmonary vasoconstrictor effects of 5-HT are mediated by the 5-HT 2A receptor and, in part, through rho kinase activation.
Researchers further demonstrated the hemodynamic effects of 5-HT infusion and 5-HT 2A receptor antagonism in a PHHN model of the ovine fetus [44][120]. As demonstrated in other animal models, prolonged constriction of the ductus arteriosus in fetal sheep leads to elevated pulmonary pressures. This elevation is augmented by 5-HT infusion and ameliorated by 5-HT 2A receptor antagonism. From this researchers conclude that 5-HT contributes to high PVR in the fetus and speculate that prolonged exposure may impact fetal pulmonary circulation and postnatal transition.
Because both TPH1 and 5-HT 2A receptor expression are increased in experimental PH and the pharmacologic blockade of the 5-HT 2A receptor is protective against the development of experimental PH, researchers hypothesized that genetic depletion of TPH1 is also protective against PH. Interestingly, this was not the case [52][127]. Contrary to researchers' hypothesis, circulating and pulmonary 5-HT is decreased in hypoxic wild-type mice compared to the controls. TPH1 knock out mice have a similar degree of hypoxia-induced alveolar simplification and pulmonary vascular remodeling as wild-type mice. Interestingly, the hypoxia-induced increase in right ventricular systolic pressures is appropriately attenuated in TPH1 knock out mice compared to wild-type mice, suggesting local pulmonary 5-HT contributes to pulmonary vascular tone. While the genetic and pharmacologic inhibitions of TPH1 have shown protection in adult models, the mixed findings in neonatal suggest inhibition may have a variable impact in the developing lung.