Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Christine Gong and Version 3 by Jason Zhu.
Sparse and conflicting evidence exists regarding the localization, expression, and regulation of phase II drug-metabolizing enzymes and drug transporters across gestational stages. To resolve the uncertainties and assumptions in current knowledge, additional pharmacokinetic (PK) data and clinical pharmacology research are required to understand drug metabolism and transport in the pregnant woman and in the placenta. This comprehensive review aims to summarize the existing knowledge of drug metabolism and transport during pregnancy, aid incorporation of enzyme and transporter changes into physiologically based pharmacokinetic (PBPK) models, and inform predictions of PK changes in the pregnant population, especially as Sponsors and Regulators seek to serve this population in drug development programs.
- pregnancy
- placenta
- gestational change
- phase II enzyme
- drug transporter
1. Introduction
Physiological changes during pregnancy affect drug pharmacokinetics (PK), including absorption, distribution, metabolism, and elimination [1]. PK changes that affect the activity of drug-metabolizing enzymes and drug transporters can differ in each pregnancy trimester [2]. These gestational changes inform the selection of safe and effective drug doses for pregnant patients and guide the decision to conduct appropriate dose monitoring during pregnancy. Approximately 81% of pregnant women take at least one prescription or over-the-counter medication during gestation, excluding vitamins and dietary supplements [3]. Despite the high prevalence of medication use during pregnancy, most medications are administered “off-label” to pregnant patients, with doses based on PK data from nonpregnant individuals [4]. With limited clinical trials conducted in the pregnant population, PK changes during pregnancy are poorly characterized and optimal dose regimens for pregnant patients are insufficiently investigated [5].
Physiologically based pharmacokinetic (PBPK) modeling is an increasingly used method for predicting drug exposure during pregnancy [5]. Utilizing mathematical equations, PBPK models incorporate known physiological changes into a mechanistic model that describes drug PK [6]. Informed pregnancy PBPK models may support the evaluation of PK data in the pregnant population, guide the proposal of safe and effective doses for clinical drug development programs, and supplement clinical pharmacology studies during regulatory approval [7]. One aspect of PBPK model predictions is informed by adequate knowledge of gestational changes in drug-metabolizing enzymes and drug transporters; for instance, knowledge of these gestational changes may inform the robust prediction of drug renal clearance, systemic exposure, and their changes across pregnancy trimesters [6]. However, due to sparse or conflicting data, the gestational changes of only a small number of phase II enzymes and drug transporters have been incorporated into PBPK modeling software. The current gaps in knowledge emphasize the need to study changes in phase II enzymes and drug transporters across gestational trimesters.
Several literature reviews have been published that examine changes in enzyme and/or transporter expression in the pregnant woman and in the placenta. Gestational changes in select phase II enzymes and renal drug transporters have been elucidated through pharmacokinetic analysis of probe drugs administered during pregnancy [8][9][8,9]. Additional evidence suggests that drug-metabolizing enzymes and drug transporters in the placenta largely affect fetal drug exposure [10], but the lack of available or consistent information regarding gestational changes in some placental transporters necessitates further research [11]. Transcription factors, steroid hormones, genetic variations, and pregnancy complications have also been observed to change the expression of placental drug transporters [12][13][12,13].
2. Placenta
The human placenta links the fetus to the mother, providing nutrients to and removing wastes from the fetal circulation [14]. In addition to its function of supporting fetal metabolism, the placenta may play a fetoprotective role by extruding xenobiotics, such as drugs, from the fetal circulation.2.1. Placental Anatomy
The human placenta possesses a hemochorial structure, in which the fetal tissue directly contacts maternal blood [15]. The fetal tissue consists of syncytiotrophoblasts, cytotrophoblasts, and vascular endothelial cells, of which the syncytiotrophoblast cells are the main barrier to drug transport [14][15][14,15]. The syncytiotrophoblasts comprise a maternal-facing brush border membrane (i.e., apical membrane) and a fetal-facing basal membrane (i.e., basolateral membrane) [14]. The apical membrane constitutes the main site of exchange for drugs, nutrients, and endogenous molecules between the maternal and fetal circulation, while the basolateral membrane provides structural attachment to cytotrophoblasts or fetal connective tissue, which houses the fetal capillaries [14][16][14,16]. Molecules are transported from the maternal uterine vasculature, across the apical and basolateral membranes of the syncytiotrophoblasts, through the fetal endothelium, and to the fetal circulation [1].2.2. Placental Drug Transport
Both passive diffusion and transporter-mediated transfer are involved in the transport of drug molecules across the placental syncytiotrophoblast [16]. The rate of passive diffusion of a drug can be affected by its molecular weight, pKa, and/or lipophilicity [17]. In general, drugs of high molecular weight demonstrate limited passive transport across the placenta. Drugs that are unionized at physiological pH tend to diffuse across the placental membrane more easily than drugs that are ionized. Passive diffusion is also a common transport mechanism for lipophilic drugs [16]. Drug transporters are membrane proteins that efflux or influx endogenous and exogenous substances [15], offering an alternative mechanism of transport for drug substrates that do not easily diffuse across the placental syncytiotrophoblast [16]. For substrates of multiple drug transporters, the net direction of transport is determined by the relative abundance of each transporter, the affinity of the drug for each transporter, and the mechanisms that regulate transporter activity [14]. The localization of select placental drug transporters and the directionality of their transport are illustrated in Figure 1. A description of each drug transporter family is detailed in subsequent sections.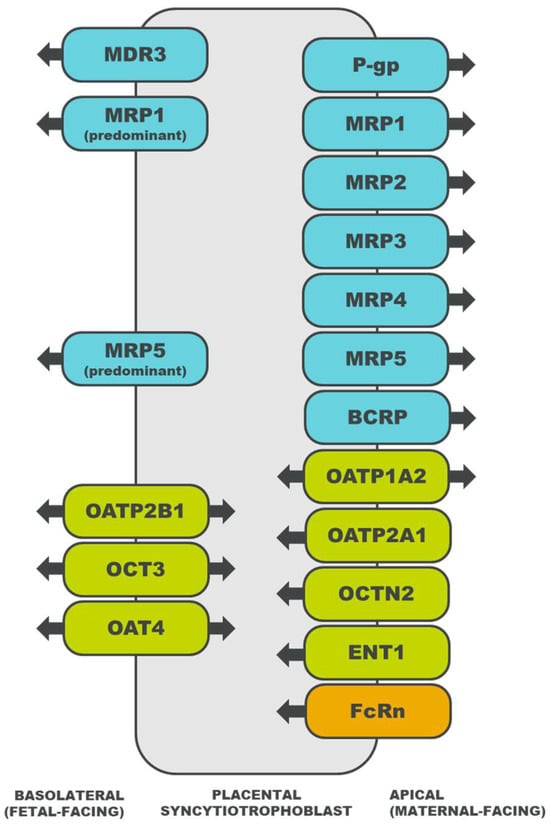
Figure 1. Localization of drug transporters in the placental syncytiotrophoblast and the directionality of their transport [18][19][20][21][22][23][24][25][26][27][28][29][30][31][32][33][34][35][36][18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36]. ATP-binding cassette (ABC) transporters are depicted in blue, solute carrier (SLC) transporters in green, and an immunoglobulin transporter in orange. MDR, multidrug resistance protein; P-gp, P-glycoprotein; MRP, multidrug resistance associated protein; BCRP, breast cancer resistance protein; OATP, organic anion transporting polypeptide; OCT, organic cation transporter; OCTN, organic cation/carnitine transporter; OAT, organic anion transporter; ENT, equilibrative nucleoside transporter; and FcRn, neonatal Fc receptor.