1. Introduction
Poly(ADP-ribose) polymerase (PARP) are a family of multifunctional enzymes involved in several cellular processes, including DNA repair mechanism and apoptosis
[1][48]. PARP-1, a nuclear, zinc-finger, DNA-binding protein that has been identified as the most abundantly expressed and characterized isoform of the PARP family, localizes to DNA strand breaks as part of the base excision repair process
[2][49]. Upon detection of the DNA damage, PARP-1 catalyzes the addition of a poly-ADP-ribose (PAR) chain to target proteins, thus recruiting repair factors to repair DNA
[3][50]. It is well known that PARP inhibitors are particularly effective against tumors carrying mutations in
BRCA. In fact, it has been demonstrated that it is possible to achieve synthetic lethality and increased tumor cell death through the prevention of DNA repair via PARP inhibition, in conjunction with the loss of broken double-strand DNA repair via
BRCA-dependent mechanisms. In other words, it has been indicated that the detection of a mutated
BRCA gene in some TNBC patients results in the blockage of the repair process of broken single-stranded DNA via PARP inhibitors by obstructing PARP enzyme activity and PARylation reactions through the competition with coenzyme, NAD+, for the interaction with the PARP catalytic domain; in addition,
BRCA mutants cannot induce homologues recombination to repair double-stranded DNA, with the consequence of synthetic lethal effects on tumor cells
[4][51]. Therefore, this makes the inhibition of PARP an attractive target for TNBC tumors that are
BRCA deficient
[5][52]. The discovery and development of PARP inhibitors began more than 50 years ago using the nicotinamide functional group
[6][53]. Later, PARP inhibitors were designed by introducing nicotinamide and benzamide functional groups into their structure in order to enable them to bind to the catalytic portion of PARPs
[7][54].
2. Conventional Chemotherapy
Conventional chemotherapy, consisting of different combinations of anthracycline, taxane, cyclophosphamide, and fluorouracil, is the mainstay of adjuvant systemic treatment for most patients with early-stage TNBC, although sometimes schedules are limited by toxicity and tumor response. At present, the 90% of drug failures in metastatic cancers is caused by the development of multidrug resistance by tumor cells. In this respect, tumor cells have given rise to the ability to survive after chemotherapeutic exposure through the employment of several mechanisms, such as APC transporters, β-tubulin III, mutations in DNA repair enzymes such as topoisomerase II and DNA mismatch repair enzymes, alterations in genes involved in apoptosis, ALDH1 and glutathione (GSH)/Glutathione-S-transferase (GST), and NF-kB signaling pathways
[8][18].
In addition to standard chemotherapy, the addition of any other drugs has been regarded as a new regimen
[9][19].
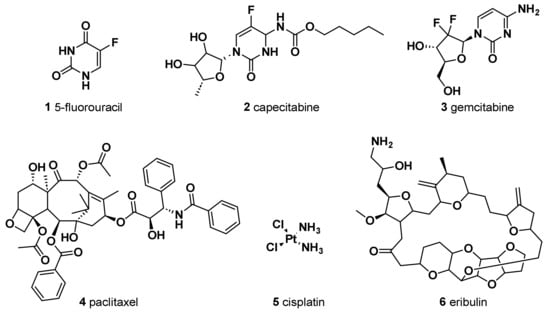
The antimetabolite 5-fluorouracil (5-FU) (
1) (
Figure 1) is widely employed in treating different cancers, including breast cancer. The chemical structure of this antitumor agent is comprised of a heterocyclic aromatic ring with high similarity to uracil, bearing a fluorine atom in 5C position of the aromatic ring. Continuous progress in comprehending its mechanism of action has led to its extensive anticancer applications
[10][20]. It has been indicated that the anticancer effect of 5-FU, when it is administered as a single agent chemotherapeutic drug, is mainly induced by its conversion to three active metabolites: fluorodeoxyuridine monophosphate (FdUMP), fluorodeoxyuridine triphosphate (FdUTP), and fluorouridine triphosphate (FUTP). In fact, these metabolites cause cell injury by two different mechanisms. Firstly, the binding of FdUMP to the nucleotide binding site of thymidylate synthase (TS), an essential enzyme for catalyzing the reductive methylation of deoxyuridine monophosphate (dUMP) to deoxythymidine monophosphate (dTMP), with the folate cofactor 5,10-methylene tetrahydrofolate (CH
2THF), as the methyl donor, resulted in the formation of a stable ternary TS–FdUMP–CH
2THF complex blocking access of dUMP to the nucleotide binding site and, therefore, in the inhibition of dTMP synthesis. Since dTMP is essential for DNA replication and repair, its depletion causes lethal DNA damage. Furthermore, the cytotoxic effect of 5-FU is also triggered by the incorporation of its active metabolite, FUTP, into RNA. This not only resulted in the inhibition of the processing of pre-rRNA into mature rRNA but also interfered with the post-transcriptional modification of tRNAs and the assembly and activity of snRNA/protein complexes, thereby inhibiting splicing of pre-mRNA. Therefore, the misincorporation of 5-FU has the potential to disrupt various facets of RNA processing, leading to significant effects on cellular metabolism and survival.
Figure 1.
Chemical structures of conventional chemotherapeutic agents.
Moreover, the outbreak of drug resistance, due to the upregulation of ATP-binding cassette (ABC) transporters, is a very common phenomenon among breast cancer patients who were administered 5-FU. In fact, it has been demonstrated that multidrug-resistant protein-1 (ABCC1/MRP1), breast cancer resistance protein (ABCG2/BCRP), and multidrug-resistant protein-8 (ABCC11/MRP8), expressed more frequently in TNBC compared to other breast cancer subtypes, are responsible for the upregulation of ABC transporters that utilize ATP to efflux various compounds—including a wide range of anticancer drugs—across cellular membranes for conferring resistance to 5-FU and other drugs that represent the backbone of current TNBC treatment. The biological effects of developing 5-FU resistance include a decline in apoptosis, disorder in cell cycle, enzyme malfunctioning, etc.
[11][21].
Capecitabine (
2) is an oral prodrug, metabolized in vivo to 5-FU by carboxylesterases, cytidine deaminase, and thymidine phosphorylase/uridine phosphorylase sequentially, which has shown effectiveness in treating advanced BC
[12][22] and gastric cancer
[13][23]. In early-stage TNBC patients, capecitabine in combination with standard adjuvant chemotherapy showed significant disease-free survival and overall better survival outcomes than standard chemotherapy with tolerable adverse events
[14][24]. A study showed that the addition of capecitabine-based chemotherapy was the most effective regime
[15][25].
Gemcitabine (2′,2′-difluorodeoxycytidine, dFdC) (
3), an efficient chemotherapeutic drug for the treatment of various types of cancer in clinical practice, was also evaluated in the treatment of TNBC in several clinical trials
[16][26]. DFdC is a potent and specific deoxycytidine analog which, once inside the malignant cell, is firstly phosphorylated by deoxycytidine kinase to its monophosphorylated form and subsequently by nucleotide kinases to its active metabolites, dFdC diphosphate (dFdCDP) and dFdC triphosphate (dFdCTP). These active metabolites are nucleosides that mediate antitumor effects. dFdCDP works as an inhibitor of ribonucleotide diphosphate reductases, an enzyme responsible for catalyzing the biosynthesis of deoxycytidine triphosphate (dCTP), which is a precursor necessary for DNA synthesis, from the corresponding ribonucleotide
[17][27]. Therefore, the overexpression of ribonucleotide reductase is associated with the emergence of resistance to dFdC
[18][28]. In addition, the cytotoxicity of dFdC is mainly associated with the cellular accumulation of dFdCTP. dFdCTP competes with dCTP for incorporation into DNA, thereby competitively inhibiting DNA chain elongation. This process is referred to as “masked DNA chain termination” and induces a G0/G1 and S-phase arrest in the cell cycle, which triggers apoptosis
[19][29].
To overcome the drug resistance induced by nucleoside kinase deficiency, various therapeutic approaches have been postulated. In this context, a multisubstrate deoxyribonucleoside kinase of
Drosophila melanogaster in the nucleus or cytosol has been reported as a potential candidate suicide gene for reversing acquired dFdC resistance in TNBC cells
[20][30].
Standard anthracycline-based chemotherapy is the treatment of choice as first-line chemotherapy for metastatic breast cancer patients not previously treated with anthracyclines
[21][31]. Its mechanism of action includes DNA intercalation, membrane binding, free radical formation, DNA repair cascade degradation, and cell death. Although anthracyclines represent an important component of adjuvant chemotherapy, they are associated with several short and long-term adverse events, with the major being cardiotoxicity and secondary leukemia
[22][23][32,33].
Nowadays, in order to avoid unnecessary anthracycline treatment due to its associated cardiotoxicity, taxane-based regimens constitute another standard therapy for breast cancer patients, especially those affected with TNBC. Paclitaxel (PTX), isolated from the bark of the Pacific Yew, and docetaxel, a semisynthetic analog from the renewable and more readily available leaves of the European yew tree, are among the most active agents for metastatic breast cancer. The assembly promoting properties of PTX (
4) were firstly reported in 1979
[24][34]. PTX, through its effects on microtubules, inhibits the growth of a variety of solid tumor cells causing, at high concentration, mitotic arrest at the G2/M phase, whereas, at low concentration, it induces cell apoptosis at the G0 and G1/S phase; however, its clinical application is limited due to poor water solubility.
Many studies have illustrated that PTX combined with 5-FU shows synergic activity and improved tolerability towards some breast carcinoma, ovarian cancer, and gastric cancer
[25][26][35,36]. Chen et al.
[27][37] have recently reported the development of KLA-modified liposomes co-loaded with 5-FU and PTX (KLA-5-FU/PTX Lps). This new drug formulation was evaluated for its antitumor activity against human breast cancer cells (MDA-MB-231). It showed enhanced cytotoxicity against MDA-MB-231 cells, improved drug delivery to mitochondria, induced mitochondria-mediated apoptosis, and turned out a promising system to target the delivery of antitumor drugs to mitochondria as a treatment for TNBC
[27][37].
For patients unresponsive to the treatment mentioned above, adjuvant capecitabine or platinum-based chemotherapy, such as carboplatin and cisplatin, might be given, although it is still controversial, as researchers are currently leading an ongoing randomized phase III trial to validate the superiority of either adjuvant capecitabine or platinum-based chemotherapy
[28][38].
A drug combination of gemcitabine and cisplatin (cis-diamminedichloroplatinum, cis-DDP (
5) has proven to be superior to dFdC/PTX in first-line treatment of metastatic TNBC in terms of progression-free survival (PFS)
[29][39]. The potential effect of nanoparticle albumin-bound (nab)-PTX and cis-DDP was assessed for metastatic mucinous adenocarcinoma
[30][40] and metastatic TNBC
[31][41]. Wang et al.
[31][41] conducted a randomized phase III controlled open-label trial to compare the efficacy of nab-PTX/cis-DDP with dFdC/cis-DDP in metastatic TNBC patients. The study confirmed that the combination of nab-PTX with cis-DDP, as compared to dFdC/PTX, significantly increased the OS and led to a significantly higher objective response rate. The reduction in the risk of death may result from the higher antitumor activity of nab-paclitaxel over gemcitabine when combined with cisplatin.
Eribulin (NSC 707389) (
6) is another medication that has been added to the armamentarium of drugs against TNBC after being studied in patients who have received at least two chemotherapeutic regimens, which include an anthracycline and a taxane. It is a non-taxane synthetic analogue of halichondrin B, isolated from the marine sponge Halichondria okadai, which acts as an irreversible inhibitor of microtubule polymerization
[32][42]. Since it does not affect the microtubule depolymerization, it causes less toxicity compared to the previously reported taxanes
[33][43]. This results into apoptosis through the disruption of mitotic spindles and an irreversible block of the cell cycle at the G2-M level. Currently, eribulin is used to treat HER2-negative metastatic or recurrent BC that failed previous exposure to anthracyclines and taxanes, and for treatment of TNBC as well
[34][44]. Eribulin in combination with the PARP-1, PARP-2, and PARP-3 inhibitor olaparib showed good efficacy for advanced TNBC patients and was well tolerated
[35][45].
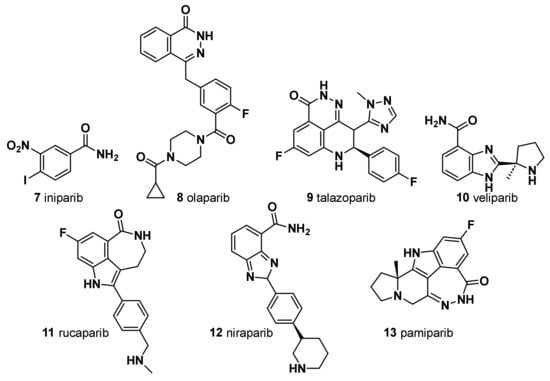
3. Iniparib (7)
Iniparib was evaluated as the first PARP inhibitor potential candidate agent for TNBC; however, it failed clinical trials and a definitive in vitro study revealed that the agent did not appreciably inhibit PARP
[36][55] (
Figure 2).
Figure 2.
Chemical structure of PARP inhibitors.
4. Olaparib (8)
Olaparib was the first small-molecule PARP inhibitor to show clinical efficacy and tolerability in
BRCA1/
BRCA2-mutated advanced BC
[37][56] (
Figure 2). First reported as a PARP-1 and PARP-2 inhibitor, it also showed a potent PARP-3 inhibition. Olaparib and talazoparib (TALA) (
9) were approved in 2018 by the Food and Drug Administration as monotherapy for the treatment of metastatic TNBC harboring a germline
BRCA1 (g
BRCA) or
BRCA2 mutation
[38][39][57,58], based on the results of two OlympiADand EMBRACA clinical trials. Approximately 15% of patients with TNBC have g
BRCA mutations, which make them good candidates as PARP inhibitors. Moreover, the use of the PARP inhibitor olaparib in the treatment of TNBC patients without
BRCA mutations has recently been shown to be ineffective
[40][59]. Noteworthy, in HER2-negative metastatic BC and
BRCA mutation patients, olaparib alone has a remarkable advantage on the standard treatment. However, cancer multidrug resistance (MDR) represents a major challenge for effective cancer treatment, and overexpression of a P-glycoprotein (P-gp) and a breast cancer resistance protein (BCRP) has been hypothesized to be one of the mechanisms responsible for acquired resistance to olaparib. Thus, a second generation of PARP inhibitors has been developed and tested in several clinical trials.
5. Talazoparib (9)
Talazoparib shows potent PARP inhibition and trapping potential superior to other PARP inhibitors
[41][60] (
Figure 2). It achieved pathologic complete responses in germline
BRCA-positive, HER2-negative patients with early breast cancer, including TNBC
[42][61]. Of note, in EMBRACA clinical trials, treatment with single-agent talazoparib in patients with advanced BC and a germline
BRCA1/
2 mutation, provided a significant PFS and a higher pathological remission rate compared to standard chemotherapy. Patient-reported outcomes were superior with talazoparib
[43][62]. Unfortunately, even with the development of a second-generation PARP inhibitor, the rise of MDR could not be prevented. Eskiler et al.
[44][63] have recently reported that talazoparib resistance is mediated by overexpression of BCRP and multidrug resistance-associated protein 1 (MRP1) genes in
BRCA1 mutant TNBC. In this regard, the authors, with the aim to overcome the abovementioned drug-resistance, developed novel talazoparib-solid lipid nanoparticles (SLNs), through a hot homogenization technique, as a promising therapeutic carrier to reverse MDR-mediated resistance in TNBC, showing significantly higher apoptotic rates in vitro than the free drug
[44][63]. In fact, several in vivo and in vitro studies reported increased intracellular drug accumulation in cancer cells and effective overcoming of drug efflux-mediated resistance of SLNs.
6. Veliparib (10)
Veliparib is a benzimidazole, having a substitution at C-4 with a carbamoyl cluster and (2
R)-2-methylpyrrolidin-2-yl moiety at C-2 (
Figure 2). In combination with carboplatin and PTX followed by doxorubicin and cyclophosphamide, it improved the pathological complete response of patients with TNBC (but not when veliparib was added to carboplatin and PTX alone). The addition of carboplatin was considered as a potential component of neoadjuvant chemotherapy for patients with high-risk TNBC
[45][64]. In addition, veliparib is being investigated in combination with radiation to treat patients with advanced TNBC, and thus far, the results are pending (NCT01618357).
7. Rucaparib (11)
A small subset of patients with high genomic loss of heterozygosity score or non-germline
BRCA1/
2 mutation derived benefit from the PARP inhibitor rucaparib
[46][65] (
Figure 2). Currently, rucaparib is under investigation in combination with other agents such as immunotherapy, vascular epidermal growth factor receptor (VEGFR) inhibitors, or radiotherapy to treat solid tumors including TNBC (NCT03992131, NCT03542175, and NCT03911453). In 2020, a study assessing the efficacy of radiotherapy in combination with rucaparib was completed, the results of which have not yet been published.
Rucaparib camsylate (camphorsulfonate salt obtained via reaction of rucaparib with one molar equivalent of (1
S,4
R)-camphorsulfonic acid) has been formulated as film-coated tablets for oral route of administration. It is indicated as a monotherapy treatment of patients with advanced ovarian cancer with germline and/or somatic
BRCA mutation who have experienced two or more chemotherapies. Rucaparib camsylate is under clinical development by Clovis Oncology and currently in phase II for TNBC
[47][66].
8. Niraparib (12)
Niraparib, in a single-arm, phase II study, demonstrated promising antitumor activity and safety in patients with localized HER2-negative,
BRCA-mutated breast cancer (
Figure 2). In a pilot, single-arm, phase II study, a neoadjuvant treatment with niraparib as a single-agent demonstrated promising antitumor activity and high levels of tumor penetration in patients with HER2-negative,
BRCA-mutated, localized BC. Niraparib showed superior tumor penetration to other PARP inhibitors
[48][67].
9. Pamiparib (BGB-290) (13)
A new PARP inhibitor undergoing clinical evaluation in patients with ovarian cancer and TNBC can inhibit PARP1 and PARP2 (NCT03333915) (
Figure 2). Moreover, it is being investigated for its efficacy in solid tumors including TNBC as a monotherapy and in combination with the chemotherapy agent temozolomide (NCT03150810). In a recent open-label, phase II, multicenter study in China (NCT03575065), pamiparib showed encouraging efficacy and an acceptable safety profile in patients with locally advanced and metastatic HER2 breast cancer with germline
BRCA1/
2 mutation
[49][68].
PARP inhibitors represent a valid therapeutical option against the TNBC (
Figure 2). Novel PARP inhibitors are currently under investigation for use alone and in combination with established agents. Although
BRCA-deficient tumors are more sensitive to PARPi due to its synthetic lethality, nearly 40% of
BRCA1/2-deficient patients do not respond to PARPi due to the emergence of drug resistance mechanisms in TNBC. It is worth noting that the administration of PTX and doxorubicin, which are substrates of the MDR1 transporter, prior to PARPi treatment may lead to the upregulation of MDR1 and indirectly induce PARP resistance. In a specific study, the use of paclitaxel before PARPi was found to be significantly linked to the presence of ABCB1 fusion transcripts
[50][69]. However, the use of doxorubicin before PARPi did not show a significant association with ABCB1 fusion transcripts. Other studies have also demonstrated the existence of cross-resistance between MDR1, PARPi, and paclitaxel
[51][52][70,71]. Therefore, conversely, the occurrence of MDR1 overexpression as a mechanism of resistance to PARPi has implications for the choice of subsequent treatment following PARPi resistance.
An even greater challenge is to decipher the mechanisms of intrinsic PARP inhibitor resistance among patients with g
BRCAm. Therefore, there is still a need to improve the therapeutic options for TNBC patients, especially in specific TN molecular subtypes
[53][72].