2. Multiple Sclerosis Therapeutics
The majority of approved treatments for MS developed to date focus on modulators of the immune system. Ocrelizumab is an intravenously administered monoclonal antibody targeting the B cell marker cluster of differentiation 20 (CD20)
[5][6]. This treatment is administered every six months with a two hour infusion. It is generally well-tolerated and highly effective for RRMS and is also one of the very few DMTs approved for treatment of PPMS. Ocrelizumab has been clinically proven to limit relapses, inflammatory activity, and disease progression in patients with RRMS. It also delays clinical and MRI progression in patients with PPMS. The relapse rate is reduced to about 46% with this medication
[5][6].
Ocrelizumab is an example of treatment which has been approved for targeting of the immune system, with the purpose of preventing initiating focal inflammatory events characteristic of relapse-remitting MS. There are a number of other approved therapeutics acting on this general process in a variety of manners, including both small molecule chemicals and antibody-based biologicals
[1]. Choosing a DMT for a patient diagnosed with MS is a challenging task. Early termination of the immune attack on the CNS with DMTs may enhance long-term clinical outcomes by minimizing neurological damage. In addition to ocrelizumab, other approved monoclonal antibodies include natalizumab, ofatumumab, and alemtuzumab, which are administered intravenously. Small molecule oral medications include ponesimod, ozanimod, siponimod, fingolimod, cladribine, dimethyl fumarate, monomethyl fumarate, diroximel fumarate, and teriflunomide, with diverse pharmacologival targets. Other non-antibody-based biologicals, such as peptides and proteins which modulate immune responses are also approved, including Glatiramer acetate, interferon-β-1b, interferon-β-1a, and pegylated interferon-β-1a
[6][7]. PPMS is the least treatable form of MS. Treatments in which the immune system is suppressed are highly effective in RRMS, but only have a modest effect in PPMS. Inflammation is persistent in all forms of MS; however, studies report that chronically active microglia and signs of neurodegeneration are more common in PPMS than RRMS. Active white matter lesions are less common in PPMS. In order to treat PPMS, the chronic activation of microglia and neurodegeneration likely needs to be targeted
[7][8].
A subset of these treatments has been demonstrated to potentially increase remyelination, which can protect against further axonal damage
[8][9]. There are a number of different potential therapeutic targets which are not yet approved for MS treatment, but which show promise in animal models, including vitamin D derivatives
[9][10]. G-protein coupled receptors (GPCRs) are also well-represented amongst these potential new targets which modulate the differentiation and activation of oligodendrocyte precursor cells
[10][11], including the endogenous opioid receptors
[11][12].
3. Kappa Opioid Receptor Agonists in Multiple Sclerosis Models
The endogenous opioid system consists of four related GPCRs, the mu (MOR), delta (DOR), and kappa (KOR) opioid receptors, as well as the nociceptin receptor (NOP)
[12][13]. This system is also comprised of endogenous opioid peptides, including β-endorphin, Met- and Leu-enkephalin, dynorphin A(1–17), and nociceptin. This system acts to modulate many physiological processes, most notably pain and reward. These functions are consistent with the system being the target of the most effective analgesic painkillers, such as oxycodone, buprenorphine, and fentanyl
[13][14]. The side effects of such opioid medications result from the extensive localization of opioid receptors throughout the body, participating in the regulation of many different functions, such as neurons of the digestive tract
[14][15]. Components of the endogenous opioid system have also been revealed in immune cells
[15][16].
The most pronounced analgesic effects of centrally active opioid receptor agonists are mediated by MOR, but both KOR and DOR-selective agonists have modest analgesic effects as well
[16][17]. In addition to analgesic functions, KOR and MOR are involved in counter-modulation of the reward system of the central nervous system (CNS)
[17][18]. Within the ventral striatum, MOR activation leads to enhanced dopamine release and KOR activation inhibits dopamine release. Consequently, whereas MOR-selective agonists such as oxycodone typically invoke euphoric effects in addition to analgesia, KOR-selective agonists such as spiradoline result in dysphoria. At low doses, the KOR-selective agonist nalfurafine results in relief for those experiencing pruritis (severe itching) without dysphoric effects
[18][19]. Nalfurafine was the first KOR agonist approved for use in humans, for the treatment of uremic pruritis in Japan
[19][20]. A second KOR agonist, the peptide difelikefalin, was more recently approved for the same indication by the Food and Drug Administration of the United States, under the brand name Korsuva. Difelikefalin is now also approved in the European Union and the United Kingdom, under the brand name Korpuvia
[20][21].
The role of the opioid system in MS has been postulated based on clinical reports, pharmacological studies, and animal models of MS
[21][22]. Investigations of the alterations of components of the endogenous opioid system in a rodent model of MS, using Theiler’s murine encephalomyelitis virus, revealed considerable decreases in spinal cord mRNA levels of MOR, DOR, and KOR
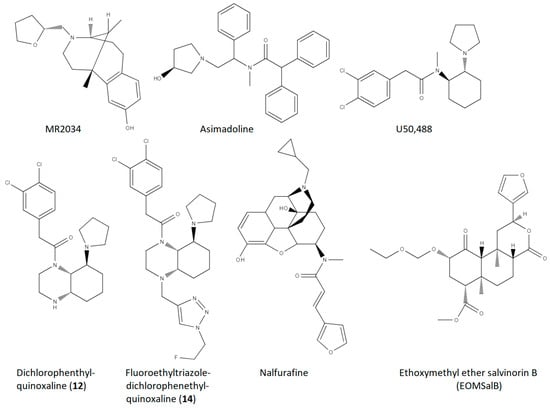
[22][23]. The first demonstration of the effectiveness of KOR ligands in ameliorating symptoms of an animal model of MS utilized the KOR agonist MR2034 (structure shown in
Figure 1) in a rat model of demyelinating disorders, the experimental autoimmune encephalomyelitis (EAE) model using rat myelin basic protein for induction
[23][24]. Typically, the EAE model involves daily assessment of animals with an ascending scoring scale involving tail limpness (1.0), hind-leg weakness (2.0), hind-leg paralysis (3.0), more extensive paralysis (tetraparesis) (4.0), and death or extreme paralysis (tetraplegia) (5.0)
[24][25]. Administration of MR2034 significantly reduced EAE scores in Dark August rats (
Table 1). Reductions were also observed in the levels of antibodies against MPB and in the pervasiveness of lesions revealed by brain histology
[23][24].
Figure 1. Molecular structures for the small molecule KOR agonist compounds which have been studied in models of MS/demyelination disorders, included in Table 1.
Table 1. Comparison of findings, and approximate magnitude of effects, in relevant published studies investigating the effects of selective KOR agonists in MS model systems. Note, in the Approximate Effect Size column, the arrows indicate the directionality of each effect. ↓ indicates a decrease in magnitude, and ↑ indicates an increase in magnitude.
| Compound |
Reference |
MS Model |
Measure |
Dose |
Approximate Effect Size |
| MR2034 |
Radulovic et al., 2010 [23][24] |
EAE, Dark August Rats |
1. EAE Score
2. anti-MBP titre
3. histological EAE lesions |
0.2 mg/kg, i.p, 11 days, 1/day |
1. 25–75% ↓
2. 40% ↓
3. 70% ↓ |
| Asimadoline |
Du et al., 2016 [25][26] |
EAE, C57BL/6 mice |
EAE Score |
5 mg/kg, i.p., 22 days, 1/day |
70–90% ↓ |
| U50,488 |
Du et al., 2016 [25][26] |
EAE, C57BL/6 mice |
1. EAE Score
2. % demyelination (MBP)
3. NG2 PBMC |
1.6 mg/kg, i.p., 22 days, 1/day |
1. 60–80% ↓
2. 60% ↓
3. 50% ↓ |
| U50,488 |
Du et al., 2016 [25][26] |
Cuprizone demyelination, C57BL/6 mice |
% remyelination |
1.6 mg/kg, i.p., 21 days, 1/day |
200% ↑ |
| U50,488 |
Du et al., 2016 [25][26] |
In vitro OPC to OL differentiation |
% MBP positive cells |
0.5 µM, 1.0 µM; 5 days |
250% ↑ |
| U50,488 |
Mei et al., 2016 [26][27] |
In vitro OPC to OL differentiation |
% MBP positive cells |
0.5 µM, 2 days |
300% ↑ |
| U50,488 |
Mei et al., 2016 [26][27] |
Lysolecithin demyelination, KOR floxed mice (control) on C57BL/6 background |
# myelinated axons in corpus callosum |
10 mg/kg, oral gavage, 10 days, 1/day |
70% ↑ |
| U50,488 |
Mei et al., 2016 [26][27] |
In vitro OPC to OL differentiation, human iPSC-derived OPC’s |
Ratio of MBP positive cells to O4 positive cells |
1.0 µM, 10 days |
250% ↑ |
| Dichlorophenethyl-quinoxaline (12) |
Tangherlini et al., 2019 [27][28] |
EAE, C57BL/6 mice |
1. EAE Score
2. % CD45+ leukocytes (CNS)
3. % IL-17+ of CD4 cells
4. % Foxp3+ Treg of CD4 cells |
2 nmol, i.p., 1–18 days |
1. 20–50% ↓
2. 80%↓
3. 60% ↓
4. 300% ↑ |
| Fluoroethyltriazole Dichlorophenethyl-quinoxaline (14) |
Tangherlini et al., 2019 [27][28] |
EAE, C57BL/6 mice |
1. EAE Score
2. % CD45+ leukocytes (CNS)
3. % IL-17+ of CD4 cells
4. % Foxp3+ Treg of CD4 cells |
2 nmol, i.p., 1–18 days |
1. 50–80% ↓
2. 90%↓
3. 70% ↓
4. 400% ↑ |
| Fluoroethyltriazole Dichlorophenethyl-quinoxaline (14) |
Tangherlini et al., 2019 [27][28] |
In vitro, human PBMC stimulation |
1. % IFN-γ of DC cells
2. % IFN-γ of T cells
3. IFN-γ levels (medium)
4. IL-10 levels (medium) |
5 µg/mL, 48 h |
1. 50% ↓
2. 70% ↓
3. 50% ↓
4. 100% ↑ |
| Nalfurafine |
Denny et al., 2021 [28][29] |
EAE, female C57BL/6 mice |
1. % mice recovered
2. days in recovery
3. # of relapses
4. % myelinated axons |
0.01 mg/kg, i.p., 1/day, 23 days (45 days for measure 4) |
1. 900% ↑
2. 2000% ↑
3. 80% ↓
4. 25% ↑ |
| U50,488 |
Denny et al., 2021 [28][29] |
EAE, female C57BL/6 mice |
1. % mice recovered
2. days in recovery
3. # of relapses |
1.6 mg/kg, i.p., 1/day, 23 days |
1. 600% ↑
2. 1500% ↑
3. modest, non-significant increase |
| Nalfurafine |
Denny et al., 2021 [28][29] |
Cuprizone, female C57BL/6 mice |
% myelinated axons, corpus callosum |
0.01 mg/kg, i.p., 1/day, 7 days |
10% ↑ (restored to healthy animal levels) |
| Ethoxymethyl ether salvinorin B (EOMSalB) |
Paton et al., 2021 [29][30] |
EAE, female C57BL/6 mice |
1. % mice recovered
2. days in recovery |
0.3 mg/kg, i.p., 1/day, 23 days |
1. 300% ↑
2. 2000% ↑ |
| U50488 |
Paton et al., 2021 [29][30] |
EAE, female C57BL/6 mice |
1. % mice recovered
2. days in recovery |
1.6 mg/kg, i.p., 1/day, 23 days |
1. 200% ↑
2. 1500% ↑ |
| EOMSalB |
Paton et al., 2021 [29][30] |
EAE, female C57BL/6 mice |
% myelinated area, cervical spinal cord |
0.3 mg/kg, i.p., 1/day, 44 days |
20% ↑ |
| U50,488 |
Paton et al., 2021 [29][30] |
EAE, female C57BL/6 mice |
% myelinated area, cervical spinal cord |
1.6 mg/kg, i.p., 1/day, 44 days |
15%↑ |
| EOMSalB |
Paton et al., 2021 [29][30] |
Cuprizone, female C57BL/6 mice |
# of GST-pi+ nuclei (corpus callosum) |
0.3 mg/kg, i.p., 1/day, 7, 14, or 21 days |
7 days—250% ↑
14 days—100% ↑
21 days—no change |
| EOMSalB |
Paton et al., 2021 [29][30] |
Cuprizone, female C57BL/6 mice |
% myelinated axons, corpus callosum |
0.3 mg/kg, i.p., 1/day, 14 or 35 days |
14 days—nonsignificant
35 days—50% ↑ |
| Cyclotide peptide– [T20K]kalata B1 |
Thell et al., 2016 [30][31] |
EAE, C57BL/6 mice |
EAE score |
10 mg/kg, i.p, one time, 7 days prior to MOG immunization |
50% ↓ |
| Cyclotide peptide– [V10K]kalata B1 |
Thell et al., 2016 [30][31] |
EAE, C57BL/6 mice |
EAE score |
10 mg/kg, i.p, one time, 7 days prior to MOG immunization |
No effect |
The finding of decreased spinal cord opioid system mRNA in the Theiler’s murine encephalomyelitis model
[22][23] were recently replicated in the mouse EAE model of MS, in which mice were immunized with myelin oligodendrocyte glycoprotein (MOG) 35–55
[25][31][26,32]. Genetic knockout studies of the KOR did not reveal overt behavioral or physiological baseline changes, but alterations in chemical pain sensitivity were noted
[32][33]. Subsequent studies investigating the effects of KOR knockout in animal models of MS demonstrated enhanced susceptibility to MS-like behaviors in the EAE model
[25][26]. Knockout of DOR also led to enhanced susceptibility in the EAE model, but the effects were more modest than those observed for KOR knockout mice. No alterations were observed for knockout of MOR. The results are consistent with the findings from the same report that oligodendrocyte precursor cells (OPCs) from C57BL/6 mice express KOR and DOR, but not MOR
[25][26].
In addition to muscle paralysis scoring, a number of neurochemical and immunological endpoints are often investigated in animal EAE models. The KOR knockout mice were demonstrated to have enhanced demyelination and enhanced leukocyte infiltration of the spinal cord compared to control treated mice, consistent with the exacerbation of clinical EAE scores
[25][26]. Administration of the KOR agonist U50,488 (
Figure 1) drastically reduced clinical EAE scores, as well as decreasing both leukocyte infiltration and demyelination. Further, U50,488 was found to significantly increase oligodendrocyte-mediated remyelination in the immune-induced demyelination model of EAE (
Table 1). An additional KOR agonist, asimadoline (
Figure 1), with substantial structural differences from U50,488, was also shown to have considerable therapeutic effects in the EAE model (
Table 1). As a model of non-immune-induced demyelination diseases, the copper–chelating toxin cuprizone is fed to mice for several weeks, and demyelination and death of oligodendrocytes ensues
[33][34]. In the cuprizone model, daily administration of U50,488 led to enhanced remyelination compared to control, with maximal effects when U50,488 was injected every day that cuprizone was fed to the animals
[25][26] (
Table 1).
In vitro oligodendrocyte remyelination studies of U50,488 demonstrated that the targeted KOR are indeed expressed by oligodendrocytes
[25][26]. The intracellular signaling mechanism downstream of KOR agonism which may mediate KOR-induced remyelination were investigated utilizing selective inhibitors of relevant intracellular signal transduction proteins. Intracellular signaling components mediating this KOR-induced remyelination were probed with select inhibitors, revealing a role for Gαi/o, L-type calcium channels and p38, but not phosphoinositide-3-kinase, protein kinase C, ERK, or JNK
[25][26]. It may be possible to achieve some degree of specificity of KOR ligands for the Gαi/o, L-type calcium channels, and p38 pathways using signaling-biased KOR agonists
[34][35][36][35,36,37].
A separate study by a different research group demonstrated similar findings of KOR agonist activity in animal models of remyelination, also utilizing the well-characterized KOR agonist U50,488
[26][27]. In this case, the choice of KOR agonist to study was guided by a systematic screen of approximately 250 GPCR-targeted compounds from a commercial library (SelleckChem). A novel high throughput remyelination assay using micropillar arrays and purified OPCs was used, and the positive “hits”, compounds which led to myelination of micropillars, were all agonists of KOR. The most effective screened compound was racemic U50,488, which was then further used for in vitro and in vivo assays (
Table 1). U50,488 was effective in restoring myelination of axons of the corpus callosum in wildtype mice. Crucially, from the standpoint of potentially translating the effectiveness of KOR agonists in rodent models of remyelination, U50,488 incubation in vitro with OPCs derived from human inducible pluripotent stem cells (iPSCs) resulted in differentiation into myelin producing oligodendrocytes
[26][27].
Given the success of the KOR agonist U50,488 in ameliorating EAE and stimulating remyelination, investigation of other KOR agonists in these models is of obvious interest, both for MS as well as other demyelinating disorders, including those with no autoimmunity aspect. An interesting series of compounds based on a novel quinoxaline-based structure developed in the laboratory of Bernhard Wünsch
[37][38] was tested in neuroinflammation and neuromodulatory markers generally, and, of particular interest, in mice using the EAE model
[27][28]. In this case, the investigators were not targeting the remyelinating aspect of KOR agonists with these compounds, as the premise in part was that the compounds would be peripherally selective, and potentially effective in blocking the initial immunological insult. Although they initially characterized several derivatives of dichlorophenethylquinoxalines, they tested two compounds, namely the parent compound with no 4N substituent (secondary amine, labeled as compound
12) and a 4N-substituted fluoroethyltriazole derivative (compound
14) (
Figure 1). In in vitro neuroinflammatory investigation of human peripheral blood mononuclear cells (PBMCs), compound
14, but not compound
12, led to decreased levels of the inflammatory biomarker interferon γ (IFN-γ), as well as increased levels of the neuroimmunomodulator interleukin-10 (IL-10). In corresponding in vitro experiments utilizing mouse-derived PBMCs, comparison of wild-type and KOR-knockout animals demonstrated that the effects of compound
14 on these endpoints was mediated by KOR, as expected.
In studies of mouse EAE, both compounds
12 and
14 were effective in reducing EAE scores as well as CNS infiltration of activated T cells
[27][28]. The effects of these compounds in the EAE model may in fact involve oligodendrocyte-mediated remyelination. It should be noted that the effects of these compounds in the EAE model are less extensive than those demonstrated for U50,488 by a different laboratory
[25][26]. A head-to-head comparison of compounds
12 and
14 with U50,488 would potentially be more revealing. The investigators speculate that the extent of U50,488 CNS penetration is much greater than for compounds
12 and
14, and the relative contribution of neuroinflammation amelioration versus remyelination stimulation may be substantially different in comparison to U50,488
[27][28] (
Table 1).
Of the KOR agonist compounds which had been published as having been tested in MS models up to 2020, none of the compounds tested had previously been investigated in humans, much less been approved as safe for human use. There are only a couple of KOR-specific agonists which are approved for human use, and thus it is very sensible to investigate these agonists in models of MS. More recently the KOR agonist, nalfurafine, which is approved for use in the treatment of pruritis in Japan, was examined for effectiveness in MS models
[28][29]. In this report, parametric studies of the EAE model were reported, determining a nalfurafine dose of 0.01 mg/kg to be optimal in reducing the physiological effects of MOG35-55 infusion in mice. Nalfurafine (or vehicle) was administered beginning on the first day post inoculation in which the animals exhibited a clinical EAE score of greater than 1, in contrast to the prior mentioned studies for U50,488 and the quinoxaline derivatives. This EAE model variation has a potentially greater translational value of the findings overall, as treatment would not begin in the clinical setting until symptoms are noted. In further accord with maximizing translational value, in addition to the percentage of experimental animals which exhibit recovery from the EAE symptoms, the investigators reported the number of days in recovery (EAE scores less than 1.0) and the number of relapses experienced (EAE scores returning above 1.0), on average, for each animal. In a head-to-head comparison with the previously characterized U50,488, nalfurafine was superior in each of these measures, and particularly in regard to relapse events (
Table 1), which was not reported in the previously discussed studies. Note, the effects of nalfurafine on EAE scores were partially reversed by pretreatment with the long-acting KOR agonist norBNI (injected once per week)
[28][29]. The improved effects of nalfurafine at a comparable point in the dose effect curve to U50,488 may be due to signaling bias differences
[34][36][35,37].
Nalfurafine treatment was also found to increase the percent of myelinated spinal cord axons
[28][29]. In a separate experiment, nalfurafine administration was initiated not on the day at which each animal showed a score of at least 1.0, but rather 10 days after each animal exhibits their peak EAE score. Under these conditions of nalfurafine treatment, recovery rates were increased compared to vehicle control (albeit to a lesser extent than described above for animals in which nalfurafine treatment was initiated beginning on the first day of EAE symptoms, well prior to the peak EAE scoring day (
Table 1)). In contrast to the findings described for the quinazoline compounds (
Table 1)
[27][28], there were no significant alterations in PBMC composition following 45 days of nalfurafine treatment in the EAE model, in comparison to vehicle. However, there was a reduction in T cell infiltration of the CNS following nalfurafine treatment in the longer-term chronic EAE model, which may contribute to the enhanced recovery rates for nalfurafine, in addition to the contribution of nalfurafine-induced enhancement of remyelination. Nalfurafine also had immunomodulatory properties, reducing IL-17 expression in T-cells in response to EAE antigen challenge. Further, in the separate demyelination model of cuprizone treatment, 7 days of nalfurafine at the end of cuprizone feeding resulted in restoration of percentage myelinated axons in the corpus callosum being restored to that of healthy controls
[28][29].
This group also investigated the effects of a different KOR agonist, ethoxymethyl ether salvinorin B (EOMSalB), an analog of salvinorin A, which is the active agent of the hallucinogenic herb Salvia divinorum
[29][30]. As with the nalfurafine studies described above, EOMSalB treatments (compared to vehicle or U50,488) were initiated in a mouse EAE model beginning on the day each animal began demonstrating symptoms, which resulted in an EAE score of at least 1.0. EOMSalB at a dose of 0.3 mg/kg had a statistically significant increase in the percentage of animals which recovered, compared to 1.6 mg/kg U50,488. The average number of days in recovery was also increased compared to vehicle treated animals, and spinal cord myelination levels were also enhanced (
Table 1). Similar to the results described for nalfurafine, EOMSalB (and U50,488) led to reductions in infiltration by immune cells generally, and CD4+ T-cells most particularly, into the CNS during the EAE experiment. Further, the response of splenocytes to an antigen challenge showed lower numbers of T cells with interferon or IL-17 expression in animals receiving EOMSalB or U50,488, indicating lowered inflammation in these animals, which can contribute to enhanced recovery in addition to effects on oligodendrocytes.
When administered during the final 7 days of cuprizone treatment, as well as the final 7 days and during the initial 7 days of recovery following cuprizone cessation, EOMSalB was effective in enhancing oligodendrocyte differentiation in the corpus callosum
[29][30]. Continued treatment for an additional 7 days (14 days post-cuprizone) did not yield significant differences compared to vehicle-treated animals. When treatment was performed for the final 7 days of cuprizone treatment and then an additional 4 weeks, there was a significant increase in the number of myelinated corpus callosum axons in animals receiving EOMSalB versus vehicle. EOMSalB also yielded the benefit of increasing weight gain following cessation of cuprizone, but it should be noted that it provided no effect on reversing cuprizone-induced pain or motor incoordination, as assessed by von Frey mechanical withdrawal threshold and balance on the horizontal bar assay. The use of behaviors in the standard cuprizone assay may not be particularly revealing with regard to therapeutic candidates, however
[29][30].
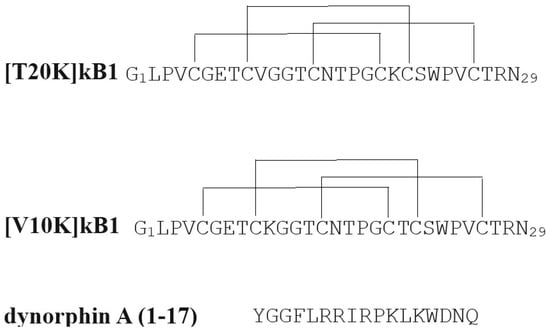
An additional putative agonist for KOR has also been revealed to have therapeutic potential in animal models of MS. A particular cyclotide, kalata B1, one of a family of natural peptides containing six conserved cysteine residues which are interlocked as three cystine disulfide bonds found in plants, was found to have immunosuppressive activities in in vitro experiments using human PBMCs
[38][39]. A single amino acid derivative of kalata B1, T20K-KB1, was shown to have significantly ameliorative effects in the EAE model of MS, whereas a different derivative, V10K-B1, was inactive in both in vitro and in vivo models (
Figure 2)
[30][31]. A subsequent study revealed T20K-KB1 to have low µM activity in inhibiting KOR radioligand binding, similarly to other select cyclotides
[39][40]. T20K-B1 also exhibited full agonism in a cAMP inhibition assay, with no effects on downstream β-arrestin signaling, indicating a G-protein-biased mechanism of action. There are no obvious sequence similarities with KOR-selective endogenous opioid peptides to explain the KOR activity of T20K-kB1 (
Figure 2, dynorphin A(1–17) sequence is shown as an example). Also, it should be noted that any interaction of the non-active cyclotide derivative V10K-kB1 with KOR was not reported upon in this second publication. Definitive determination of KOR mediation of the MS-ameliorative effects of T20K-B1 will require further investigation. Kalata B1 has other effects beyond KOR agonism, including ion-permeable multimeric pore formation in cell membranes, at concentrations which suggest this mechanism may also be relevant to the in vivo and in vitro effects of T20K-B1
[40][41].
Figure 2. Amino acid sequences of the cyclotide analogs investigated for EAE activity, as well as, in the case of [T20K]kB1, KOR activity. The amino acid sequence of an endogenous opioid peptide, dynorphin A(1–17), which is selective for KOR is shown for comparison.
4. Mechanism for KOR Agonist Effects on MS Models and Remyelination
A key facet of MS is destruction of the myelin coating of neurons—remyelination is an important factor which can help prevent recurrence of symptoms. Oligodendrocytes are the cells which produce myelin and are thus a key target for remyelination of neurons
[41][42]. The endogenous KOR system has recently been demonstrated to play a modulatory role in oligodendrocyte-mediated myelin repair (or remyelination)
[42][43]. The targeting of endogenous oligodendrocytes to effect myelin repair in MS is a tantalizing possibility for improved treatment outcomes
[43][44]. The understanding of the direct effect of most currently approved pharmacotherapeutic treatments of MS is through suppressing immune responses which are thought to be the primary incidental factor. For instance, the small molecule fumarates are thought to modulate the immune response, but they also may have modest effects on remyelination which contribute to the treatment effectiveness
[44][45][45,46].
In an in vitro enzymatic model of remyelination, neurotrophic factors which interact with the gp130/Janus kinase pathway, notably leukemia inhibitory factor (LIF) and cerebral dopamine neurotrophic factor (CDNF), were demonstrated to effect oligodendrocyte-induced remyelination. Other neurotrophic factors which are independent of this pathway, including brain-derived neurotrophic factor (BDNF), neurotrophin 3 (NT-3), and glial cell-derived neurotrophic factor (GDNF), had no remyelinating effect
[46][47]. Investigations of whether there are interactions between the gp130/Janus kinase pathway and the endogenous KOR system could reveal whether the remyelination effects of these two targets are synergistic and/or interdependent.
During pregnancy in females with MS, a tendency towards remission from recurrences has been observed, which is no longer seen following childbirth
[47][48]. As pregnancy has been revealed to result in alleviation of MS symptoms, investigators hypothesized that the hormone prolactin may be mediating this pregnancy-induced effects and found that prolactin indeed leads to remyelination in animal models
[48][49]. The positive effects of prolactin on myelination may underlie findings of increased number of childbirths in females leading to more favorable white matter trajectories
[48][49][49,50]. It may also underlie findings that in females with secondary progressive MS treated with domperidone, subjects with higher prolactin levels had reduced disease progression
[50][51]. Prolactin may be generally protective of white matter health
[51][52], and elevated circulating levels of prolactin during sleep have been proposed as potentially mediating the enhanced levels of remyelination during sleep compared to during wakefulness
[52][53][53,54]. One of the well-known effects of KOR agonists in vivo is to cause significant and reliable increases in circulating prolactin following release from pituitary lactotroph cells. It would be of interest to investigate if the remyelination in response to KOR agonists might be mediated, in whole or in part, by the release of prolactin. It should be noted that mouse models of MS do not show appreciable effects of prolactin or prolactin receptor knockout
[54][55].
Sex differences in both MS proclivity and in the endogenous KOR system have been documented, but whether these are related has not yet been determined. The effectiveness of sex hormones in treating MS
[55][56], however, has provocative implications for the mechanisms underlying MS susceptibility, which warrant further investigation.
5. Considerations for KOR Agonists as Pharmacotherapeutics
MOR agonists have been the predominant analgesic pharmacotherapeutics since long before the endogenous opioid system was even characterized. Compounds specific to a related endogenous opioid target were only identified much later (both KOR and DOR). KOR agonists have been proposed as potential pharmacotherapeutics for a range of conditions, including peripheral pain states, itching (pruritis), and substance use disorders, among others
[56][57].
The KOR agonists (
Figure 1) which have thus far been investigated and published in KOR models, are but a small subset of a rich pharmacopeia of KOR-selective agonists
[56][57]. MR2034 was an early derivative of the benzomorphan compound ketocyclazocine, for which the kappa opioid receptor was originally named, the Greek letter for the ‘k’ in ketocyclazocine (similar to mu for morphine in the mu opioid receptor)
[57][59]. A structurally distinct agonist, U50,488, was developed by the Upjohn Company in the early 80s as an arylacetamide, with promise as a potential peripheral analgesic
[58][60]. An analog of U50,488, spiradoline (U62,066), was found to induce sedation and dysphoria
[59][61]. Asimadoline, an arylacetamide derivative as well, was developed as a more peripherally selective KOR agonist with the potential to demonstrate fewer side effects
[60][62]. Similarly, the phenethylquinoxaline compounds featured here,
12 and 14 from
[27][28], were developed as relatively peripherally selective analogs of a novel highly KOR-selective scaffold that combined the structural elements of U50,488 and a piperidine containing KOR agonist, GR89696
[61][63]. As can be seen from
Figure 1, nalfurafine has a disparate structure from the arylacetamide-containing ligands, having been developed based on the general opioid receptor binding by morphinan compounds, but with a KOR “message” moiety on the 6 Carbon
[62][64]. EOMSalB, a derivative of the chief metabolite of the primary bioactive compound present in the hallucinogenic herb Salvia Divinorum
[63][65], was found from screening the KOR activity of analogs prepared using protecting groups on salvinorin B, and determined to be among the most selective and potent KOR agonists.
Although a subset of KOR agonists is expected to yield significant negative side effects such as hallucinations, sedation, and anhedonia, at least at high doses, the use of G-protein signaling-biased KOR agonists has the potential to avert these problematic issues
[35][36][36,37]. Indeed, nalfurafine itself has been reported to be G-protein biased
[34][64][35,66]. A comparison of the G-protein-biased nalfurafine to the arrestin signaling-biased KOR agonist WMS-X500 suggest specific conformational changes amongst the fifth and sixth transmembrane domain of KOR
[64][66]; contribution of these effects and differential effectiveness in KOR agonist amelioration of MS symptoms remains to be investigated.
As mentioned above, the study by Du et al. presented the results of in vitro mechanistic studies of KOR signal transduction elements mediating U50,488-induced myelination initiation by oligodendrocytes
[25][26]. Investigating other KOR agonists in such mechanistic studies in addition to animal remyelination models may help reveal common elements to the therapeutic effects of diverse KOR agonists, and potentially aid in selection of the optimal KOR agonists for development towards MS therapeutic agents. Although the results of published studies, as summarized in
Table 1, uniformly and consistently suggest KOR to be a promising target for MS treatment, the plethora of known KOR agonists are unlikely to be equally effective, and comparative mechanistic studies could be a crucial guide in selecting optimal KOR agonists for development. The evaluation of which of the KOR agonist compounds which have thus far been tested in demyelination models (for which
Table 1 is currently a comprehensive list of all published studies) is likely to be most promising will benefit from just such in vitro mechanistic studies. KOR agonists have some positive effects leading to enhanced remyelination in both the EAE and cuprizone models of MS
[25][26][27][28][29][30][42][26,27,28,29,30,31,43]. The optimal dosing pattern which leads to the remission in MS symptoms and its relation to remyelination is still an area for investigation. Whether once-daily, steady-state KOR activation, compared to episodic use following MS recurrence, is optimal is not yet known. The consistency of the KOR agonists in ameliorating MS-like symptoms in animal models suggests that most if not all of the myriad of KOR agonist compounds could be candidates. Additional mechanistic, pharmacokinetic, and toxicological/side-effect studies will be of immense benefit to any such evaluation.
The majority of the KOR agonists which have thus far demonstrated efficacy in animal demyelination models are not approved for clinical use. Testing these compounds for effectiveness in MS in humans would require considerable further development to first get approval for these compounds as Investigational New Drugs (IND)
[65][67]. These studies involve considerable in vivo and in vitro investigations into each compound’s toxicity, pharmacokinetics, absorption, excretion, and metabolism. Also, contracting to synthesize the compound according to the Good Manufacturing Process is required prior to approval. Following IND approval, phase 1 clinical trials showing safety in healthy volunteers are typical, prior to investigations of effectiveness in disease progression (phase 2), which would then need to be followed by larger clinical trials. This somewhat daunting process typically results in many years between demonstration of effectiveness in animal models of disease and approved medications. However, as mentioned previously, two KOR agonists have been approved for clinical use by at least one governmental agency for the treatment of pruritis, nalfurafine and difelikefalin. difelikefalin has not yet been evaluated in animal or cellular demyelination models. Given that it is peripherally selective, there is the possibility that the effects would be considerably different in vivo compared to centrally active agonists. Nalfurafine has an established safety record in humans and might therefore potentially be advanced into studies much more rapidly. Further, nalfurafine is effective in treating pruritis at doses which are not aversive or sedating
[66][68]; whether this would also be true for effective doses for MS amelioration would need to be established.