Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Peter Tang and Version 1 by Susana P. Pereira.
Chronic diseases represent one of the major causes of death worldwide. It has been suggested that pregnancy-related conditions, such as gestational diabetes mellitus (GDM), maternal obesity (MO), and intra-uterine growth restriction (IUGR) induce an adverse intrauterine environment, increasing the offspring’s predisposition to chronic diseases later in life. There are several CD risks factors an individual can manage. Recent research suggested that the intrauterine environment, which is modulated by maternal behaviors and disease, severely influences the offspring’s CD development risk.
- maternal antioxidant supplementation
- disease prevention
- chronic diseases
- developmental programming
- metabolic dysfunction
- oxidative stress
1. Introduction
Non-communicable chronic diseases (CD) are considered to be one of the major threats to global health and include cardiovascular disease, diabetes, obesity, cancer, chronic respiratory diseases, and chronic liver disease [1]. It is estimated that, in 2016, non-communicable CD contributed to two-thirds of mortality worldwide [2]. There are several CD risks factors an individual can manage, including high blood pressure, tobacco smoking, high body mass index, physical inactivity, and constant consumption of poor diets [1]. In addition, unmanageable risk factors can also contribute to CD development, such as age, sex, and genetic background [3]. On top of that, recent research has suggested that the intrauterine environment, which is modulated by maternal behaviors and disease, including gestational diabetes mellitus (GDM) [4[4][5],5], maternal obesity (MO) [5,6[5][6][7][8],7,8], and IUGR [9], severely influences the offspring’s CD development risk.
Gestational diabetes mellitus (GDM) is defined as hyperglycemic and glucose intolerance states that are detected, for the first time, either in the second or third trimester of pregnancy [10]. It is well established that GDM is the most prevalent pregnancy complication, affecting 13.9% of pregnancies [4,11][4][11]. Identified risk factors for GDM development encompass maternal age and obesity, with obese pregnant women presenting a 2.4-fold higher risk of developing GDM [12]. Furthermore, MO itself represents a highly prevalent pregnancy complication [5]. It is estimated that approximately 50% of pregnancies occur in overweight or obese women [6]. Both GDM and MO contribute to an increased risk of inducing a fetoplacental environment resembling prolonged hypoxia, along with other factors, such as maternal smoking, vascular dysfunction, and maternal nutrient reduction [13]. These conditions can potentiate suboptimal fetal development and growth, a condition referred to as intra-uterine growth restriction (IUGR). Indeed, neonatal complications associated with GDM, MO, and IUGR include increased perinatal mortality and morbidity, deviations in birthweight, and preterm birth [14,15,16][14][15][16]. These pregnancy-associated disorders induce structural, functional, and metabolic adaptations across several organs as early as the fetal stage. In this context, it has been pointed out that oxidative stress and mitochondrial dysfunction may be pivotal mechanisms of developmental programming in pregnancy-related disorders [7]. Therefore, these mechanisms can be strategic targets to modulate the programming of non-communicable CD in offspring during pregnancy.
2. Developmental Programming of Chronic Diseases by Pregnancy-Associated Disorders
2.1. (Patho)physiologic Role of Reactive Oxygen Species in Fetal Development
Proper fetal development hinges on an interplay of several critical factors, among which a consistent and uninterrupted provision of nutrients and oxygen plays a pivotal role [17]. Oxygen levels exhibit a fine orchestration throughout gestation according to the specific requirements of the developing fetus [18]. In the early stages, oxygen is maintained at lower levels, particularly up until the 12th week following conception [19]. This intentionally lowered oxygen levels stimulate angiogenesis, promoting the formation of new blood vessels, which is a vital process for sustaining early development [19]. Around the 16th week of gestation, a significant shift occurs, with intrauterine oxygen levels increasing significantly and then remaining stable until birth [19]. Even in this carefully regulated environment, a small fraction of the oxygen required for oxidative metabolism undergoes incomplete reduction, giving rise to reactive oxygen species (ROS) [19]. ROS production predominantly occurs via the escape of electrons from the mitochondrial electron transport chain (ETC), with complex-I and -III being notable contributors, resulting in the formation of the superoxide (•O2−) radical. Notwithstanding, other sources of ROS production can be considered, such as dihydroorotate dehydrogenase (DHODH), among others. It is important to state that ROS have a biphasic effect [20]. On the one hand, at moderate levels, ROS are key players in pregnancy physiology, acting as signaling molecules in developmental processes including placental growth [21], embryo development, and implantation [22], and are involved in the replication, differentiation, and maturation of cells and organs. However, on the flip side, excessive ROS levels, when not counterbalanced by the antioxidant capacity, can usher in oxidative stress [5]. This state of oxidative stress can inflict severe damage, compromising the structural integrity of cells and organelles’ membranes and hindering proper protein function. Furthermore, it poses a significant risk to fetal development and the intricate process of proper organogenesis. In sum, the orchestration of oxygen levels and the balance of ROS levels within the maternal–fetal interface are pivotal to ensuring the proper progression of fetal development.2.2. The Impact of Pregnancy-Associated Disorders on Offspring’s Organs Oxidative Stress and Mitochondrial Function
Pregnancy is a state of high energetic demand that is sustained mainly by fetoplacental metabolic activity [23]. Mitochondria are metabolism’s key players and one of the main cellular energy sources [24], being highly responsive organelles to energy demands. Consequently, placental mitochondrial function can be modulated [24] in response to an adverse intra-uterine environment. In pregnancy-related disorders, placental metabolic dysfunction has been extensively documented, which may translate into long-lasting consequences for the offspring in the prenatal and postnatal periods. This section aims to discuss how GDM, MO, and IUGR are related with fetoplacental dysfunction and offspring organ dysfunction via mitochondrial malfunction and oxidative stress. The characteristic hyperglycemic state during GDM may adversely impact the placental mitochondrial structure and function [25] (Figure 1). Indeed, placentas from GDM-portraying women presented swollen and disrupted mitochondria, some of which were completely damaged [26]. Since mitochondria are highly dynamic organelles, changing the number, morphology, network, and size according to cellular energy needs, mitochondrial shape and function are tightly linked [27]. For instance, GDM-derived human cytotrophoblasts present a decreased mitochondrial maximum respiratory capacity and decreased ATP production rates [28[28][29],29], highlighting GDM-induced mitochondrial bioenergetic dysfunction (Figure 1). Maternal diabetes, either pregestational type 2 diabetes or GDM, impair placental mitochondrial biogenesis (formation of new mitochondria), with decreased mitochondrial transcription factor A (TFAM) [30] and peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC1-α) expression levels [28,29][28][29]. In addition to impaired placental mitochondrial biogenesis, GDM leads to altered mitochondrial dynamics, with studies in humans reporting either an increase in placental mitochondrial fusion events [28,31][28][31] or a decrease [32]. Fusion events have been suggested as essential for mitochondrial DNA (mtDNA) copy number maintenance [33] (Figure 1). Although inconsistent, mtDNA copy number alterations are reported in GDM-related studies, either in maternal serum [34,35][34][35] or placental tissue [28,30][28][30]. A potential direct relationship between the mtDNA copy number and oxidative stress has been suggested [36]. This hypothesis was raised because the placental mtDNA copy number was positively correlated with placental DNA oxidation [36] both in GDM and control pregnancies. Further studies are required to understand the mechanisms linking DNA oxidation and the mtDNA copy number to solidify this hypothesis. Despite this, placental oxidative stress has been widely reported in GDM placentas. Although ROS are generated from several sources, mitochondria are considered as one of the major ones [5]. GDM human pregnancies present placental increased biomarkers of oxidative stress, such as malondialdehyde (MDA) [37[37][38],38], reduced antioxidant defenses (decreased catalase (CAT) activity [38], and glutathione peroxidase (GPx) 1 [39]) (Figure 1). In addition to the placenta, GDM human umbilical cord mesenchymal stem cells (hUC-MSC) present increased ROS production, detected via 2′,7′-Dichlorofluorescin diacetate (DCFDA), and impaired mitochondrial bioenergetics, including a diminished basal respiration state and FCCP-induced maximum respiratory capacity [40]. Given that mesenchymal stem cells (MSC) are multipotent, they can differentiate into a wide range of cell types during fetal development, including adipocytes, cardiomyocytes, myocytes, and neurons [41]. Dysfunctional MSC may imprint dysfunctional cells and organs in the offspring that may later lead to an increased CD risk.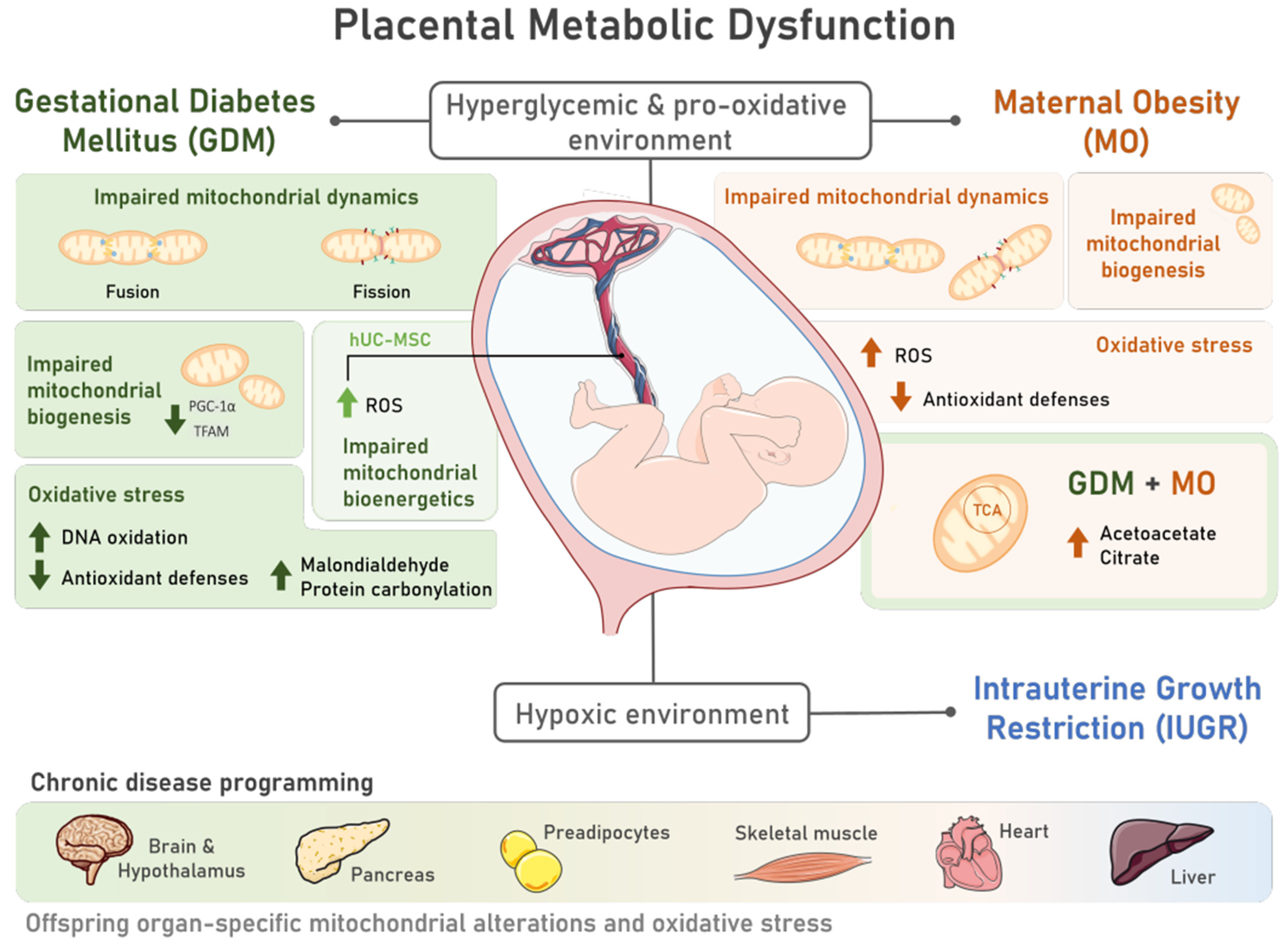
Figure 1. Placental metabolic dysfunction during pregnancies characterized by the presence of Gestational Diabetes Mellitus (GDM) and Maternal Obesity (MO) includes mitochondrial structural and functional alterations and oxidative stress, contributing to offspring chronic disease programming. The characteristic hyperglycemic and pro-oxidative intrauterine environment of pregnancies complicated by GDM and MO induces placental metabolic dysfunction via alterations in mitochondrial dynamics, with unbalanced fission and fusion events; mitochondrial biogenesis, via decreased peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α) and mitochondrial transcription factor A (TFAM) levels; and impaired mitochondrial bioenergetics, which was also observed in multipotent human umbilical cord mesenchymal stem cells (hUC-MSC) of GDM pregnancies. Increased reactive oxygen species (ROS) are also common to both placenta and hUC-MSC tissues in both pregnancy disorders. Placental oxidative stress is further marked by reduced antioxidant defenses and increased DNA oxidation, protein carbonylation, and malondialdehyde levels in GDM placentas. MO during a GDM pregnancy may further contribute to mitochondrial dysfunction, as increased levels of metabolites involved in the tricarboxylic acid (TCA) cycle and ketogenesis (citrate and acetoacetate, respectively) were detected. This compromised intrauterine environment can progress into a hypoxic state, increasing the risk of Intrauterine Growth Restriction (IUGR) development. GDM, MO, and IUGR have been associated with an increased risk of offspring chronic disease. Mitochondrial alterations and oxidative stress, which are intimately involved in chronic diseases, were observed in human and animal studies in different tissues of GDM and MO offspring, such as the brain and hypothalamus, pancreas, preadipocytes, skeletal muscle, heart, and liver, highlighting the role of metabolic pregnancy disorders in disease programming.
3. Antioxidants and Mitochondriotropic Activity of Naturally Occurring and Synthetic Antioxidants
Developmental programming due to MO, GDM, and IUGR involves a complex interplay of tissue-specific and general mechanisms [82][57], in which epigenetic changes, mitochondrial changes, and oxidative stress are highlighted as the main players in this relationship [7]. Moreover, tissue and mitochondrial dysfunction commonly resulting from an exacerbation of pregnancy-associated oxidative stress results in maternal metabolic dysfunction in a positive feedback loop [83][58]. Thus, maternal supplementation with antioxidants represents a promising approach to mitigate developmental programming effects in the offspring exposed to pregnancy-related disorders characterized by elevated ROS. Antioxidant therapy has been studied as a possible strategy for exogenous antioxidants, such as those from dietary and synthetic sources, to act synergistically with endogenous antioxidants in restoring redox homeostasis [84][59].3.1. Naturally Occurring Antioxidants
3.1.1. Endogenous Antioxidants
Under physiological conditions, the human body naturally produces antioxidant molecules to eliminate excessive ROS, preventing oxidative stress and subsequent tissue damage [85][60]. The endogenous antioxidant defense system contains antioxidant enzymes such as SOD, CAT, and GPx, as well as non-enzymatic compounds such as glutathione, proteins like ferritin, and low molecular weight scavengers such as Coenzyme Q10 (CoQ10) that are responsible for maintaining cellular redox homeostasis [86][61]. SOD belongs to the first line of the antioxidant defense system by converting the superoxide anion radical to hydrogen peroxide (H2O2) [85,86][60][61]. In turn, both CAT and GPx are involved in the reduction of H2O2 to water and molecular oxygen [85,86][60][61]. In fact, enzymatic antioxidants are more effective in counteracting oxidative stress due to their ability to eliminate ROS, preventing damage to proteins, DNA, and lipids [85][60]. On the other hand, non-enzymatic antioxidants can act by capturing transition metal ions that are mostly responsible for producing reactive oxygen radical species or can store metal ions necessary for the synthesis of enzymes containing metal ions (metal-binding proteins, e.g., ferritin and transferrin) [85,86][60][61]. Moreover, they can scavenge reactive radicals (e.g., glutathione and uric acid) and directly interfere with the initiation and propagation steps of the peroxidation process, acting as inhibitors of lipid peroxidation [85,86,87][60][61][62] and protecting cells from harm, playing an important role in metabolism as is the case of CoQ10, a naturally occurring endogenous lipid-soluble antioxidant located in the inner mitochondrial membrane.3.1.2. Dietary Antioxidants
Dietary antioxidants such as Vitamin C and E, carotenoids, and polyphenols are naturally occurring exogenous antioxidants that complement the activity of the endogenous antioxidant defense system [86][61]. Research has been dedicated to unraveling the mechanisms underlying the actions of dietary phytochemicals, with a particular emphasis on polyphenols, and how polyphenols exert their beneficial effects, including their antioxidant capacity. Polyphenols are subdivided into two major classes: flavonoids, which include quercetin and epigallocatechin-3-gallate (EGCG), and non-flavonoids, which include resveratrol and curcumin [88][63]. Dietary polyphenols present multiple benefits to human health, such as anti-cancer [89][64], antioxidant [90[65][66],91], anti-inflammatory [91[66][67],92], anti-obesity [91][66], and anti-diabetic [93][68]. Among the natural polyphenols, the most extensively researched compounds are resveratrol (3,4′,5-Trihydroxystilbene), curcumin (1E,6E)-1,7-bis(4-hydroxy-3-methoxyphenyl)hepta-1,6-diene-3,5-dione), EGCG, and quercetin, owing to their impact in biological properties. Polyphenols have been considered a potential therapeutic approach due to their ability to mitigate oxidative stress events via ROS scavenging, modulating ROS-removing enzymes, inhibiting ROS-producing enzymes, or via the chelation of metals that are involved in metal-dependent hydroxyl formation via the Fenton reaction [88][63]. Although polyphenols have the capacity to reduce ROS, which is intrinsically related to their chemical structure and the presence of at least one phenol group, the indirect action of dietary polyphenols relies on the upregulation of endogenous antioxidant proteins [85][60]. It has been suggested that resveratrol activates SIRT1 by directly inhibiting phosphodiesterase (PDE) enzymes, increasing cAMP levels and AMPK activation with a subsequent increase in NAD+ levels activating SIRT1 [94][69]. SIRT1 activation is associated with a multitude of beneficial effects, namely the ability to decrease oxidative stress, inflammation, and regulation of the expression of mitochondrial genes involved in biogenesis and lipid metabolism (SIRT1/PGC-1α axis) [95,96,97,98,99,100,101,102,103,104][70][71][72][73][74][75][76][77][78][79]. Although the mechanism is not yet well known for curcumin’s action on SIRT1, this protein is also suggested to represent a target of curcumin. Additionally, research has suggested that curcumin’s antioxidant action also relies on its ROS scavenging properties, which are attributable to the compound’s structure. On top of the mechanisms of action described above, an important indirect mechanism to mitigate cellular oxidative stress is the activation of the nuclear factor erythroid 2–related factor 2 (Nrf2)/antioxidant responsive element (ARE) pathway. Both resveratrol [105][80] and curcumin [106,107,108][81][82][83] can induce the transcriptional activation of Nrf2 and subsequent upregulation of the expression of Nrf2 target genes, including NAD(P)H:quinone oxidoreductase 1 (NQO1) and heme oxygenase-1 (HO-1), conferring an additional antioxidant ability by increasing the expression of antioxidant enzymes. In addition to the mitigation of oxidative stress, antioxidant compounds present effects in other biological processes, such as mitochondrial function and inflammation. For instance, resveratrol has been reported to induce BNIP3-related mitophagy and attenuate hyperlipidemia-related endothelial dysfunction [109][84] via the involvement of AMPK and hypoxia-inducible factor 1 (HIF-1). On the other hand, resveratrol effects can also contribute to increasing mitochondrial activity by regulating the expression of genes associated with oxidative phosphorylation, biogenesis, and lipid metabolism [95,97][70][72]. Curcumin’s main described mechanisms of action in mitochondria are targeting mitochondrial biogenesis, intrinsic apoptosis, mitochondrial permeability transition pore, mitochondrial MRC uncoupling, and ATP synthase [88][63]. Resveratrol and curcumin have also been associated with potential anti-inflammatory mechanisms. These compounds have demonstrated the ability to inhibit the nuclear factor-kB (NF-kB) signaling pathway, which subsequently prevents the expression of inflammatory cytokines such as tumor necrosis factor-alpha (TNF-α) and interleukin-6 (IL-6) (resveratrol [110[85][86],111], curcumin [112][87]).3.2. Synthetic Antioxidants
In addition to naturally occurring antioxidants, synthetic antioxidant molecules such as N-Acetylcysteine (NAC), MitoTEMPO, and Mitoquinone (MitoQ) possess the capability to exert direct and indirect antioxidant effects in vivo [84,113,114][59][88][89]. These compounds have been developed to overcome mainly pharmacological limitations. For instance, NAC, a cysteine derivative, overcomes the cysteine low intracellular concentration limits, contributing to a more efficient rate of GSH synthesis [115,116][90][91]. Therefore, NAC not only makes a significant antioxidant contribution by serving as a precursor to intracellular GSH [116[91][92],117], but also directly interacts with certain free radicals and activates the Nrf2 signaling pathway, providing an effective means to combat oxidative stress at the cellular level [118][93]. In turn, as mitochondrial oxidative stress is associated with multiple diseases, targeting specifically mitochondrial ROS and modulating redox signaling may represent a more efficient antioxidant therapy [119][94]. However, it is still a challenge to specifically target and accumulate antioxidant molecules within the mitochondrial matrix [119][94]. MitoTEMPO and MitoQ are some examples of successful mitochondria-targeted antioxidants [119][94].4. Exploring the Role of Dietary Antioxidant Supplementation during Pregnancy in Offspring Non-Communicable Disease Prevention
Maternal nutritional status is of paramount relevance during the pre- and post-conception period and throughout lactation for optimal fetal development [130][95]. Evidence suggests that maternal dietary adjustments, such as maintaining a balanced energy and protein intake, can be beneficial during these phases to reduce the risk of fetal loss, stillbirth, and perinatal death [131][96]. Although a maternal high-protein diet has been associated with a higher percentage of small for gestational age (SGA) infants [131][96], the supplementation of specific amino acids has shown positive outcomes. Specifically, oral L-Arginine supplementation during pregnancy has been shown to improve fetoplacental circulation and birth weight in humans [132][97] and increase fetal viability and birth weight in pigs and sheep [132][97]. In a non-pregnant individual’s liver, it has been reported that L-Arginine can induce antioxidant response via stimulation of GSH synthesis and activation of the Nrf2 pathway, highlighting the potential role of L-Arginine to modulate antioxidant defenses [133][98]. Moreover, antenatal iron and folic acid supplementation are recommended by the WHO to prevent fetal and neonatal loss, SGA, maternal anemia, and iron deficiency [134][99]. In non-pregnant individuals with iron deficiency, the supplementation led to a significant decrease in oxidative stress [135][100], although it remains controversial regarding maternal supplementation, particularly in already-iron-sufficient mothers [136][101]. The overall diet’s antioxidant impact may vary, ourthe main focus in this reviewsearch is centered on specific antioxidant supplementation in pregnancy-associated disorders and its potential to mitigate offspring’s development of CDs.References
- Bauer, U.E.; Briss, P.A.; Goodman, R.A.; Bowman, B.A. Prevention of Chronic Disease in the 21st Century: Elimination of the Leading Preventable Causes of Premature Death and Disability in the USA. Lancet 2014, 384, 45–52.
- Nugent, R. Preventing and Managing Chronic Diseases. BMJ 2019, 364, l459.
- Adams, M.L.; Grandpre, J.; Katz, D.L.; Shenson, D. The Impact of Key Modifiable Risk Factors on Leading Chronic Conditions. Prev. Med. 2019, 120, 113–118.
- Tocantins, C.; Diniz, M.S.; Grilo, L.F.; Pereira, S.P. The Birth of Cardiac Disease: Mechanisms Linking Gestational Diabetes Mellitus and Early Onset of Cardiovascular Disease in Offspring. WIREs Mech. Dis. 2022, 14, e1555.
- Diniz, M.S.; Hiden, U.; Falcão-Pires, I.; Oliveira, P.J.; Sobrevia, L.; Pereira, S.P. Fetoplacental Endothelial Dysfunction in Gestational Diabetes Mellitus and Maternal Obesity: A Potential Threat for Programming Cardiovascular Disease. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2023, 1869, 166834.
- Diniz, M.S.; Grilo, L.F.; Tocantins, C.; Falcão-Pires, I.; Pereira, S.P. Made in the Womb: Maternal Programming of Offspring Cardiovascular Function by an Obesogenic Womb. Metabolites 2023, 13, 845.
- Grilo, L.F.; Tocantins, C.; Diniz, M.S.; Gomes, R.M.; Oliveira, P.J.; Matafome, P.; Pereira, S.P. Metabolic Disease Programming: From Mitochondria to Epigenetics, Glucocorticoid Signalling and Beyond. Eur. J. Clin. Investig. 2021, 51, e13625.
- Pereira, S.P.; Grilo, L.F.; Tavares, R.S.; Gomes, R.M.; Ramalho-Santos, J.; Ozanne, S.E.; Matafome, P. Programming of Early Aging. In Aging; Elsevier: Amsterdam, The Netherlands, 2023; pp. 407–431.
- Holemans, K.; Aerts, L.; Van Assche, F.A. Fetal Growth Restriction and Consequences for the Offspring in Animal Models. J. Soc. Gynecol. Investig. 2003, 10, 392–399.
- Ye, W.; Luo, C.; Huang, J.; Li, C.; Liu, Z.; Liu, F. Gestational Diabetes Mellitus and Adverse Pregnancy Outcomes: Systematic Review and Meta-Analysis. BMJ 2022, 377, e067946.
- Moon, J.H.; Jang, H.C. Gestational Diabetes Mellitus: Diagnostic Approaches and Maternal-Offspring Complications. Diabetes Metab. J. 2022, 46, 3–14.
- Kim, S.Y.; England, L.; Wilson, H.G.; Bish, C.; Satten, G.A.; Dietz, P. Percentage of Gestational Diabetes Mellitus Attributable to Overweight and Obesity. Am. J. Public Health 2010, 100, 1047–1052.
- Li, H.P.; Chen, X.; Li, M.Q. Gestational Diabetes Induces Chronic Hypoxia Stress and Excessive Inflammatory Response in Murine Placenta. Int. J. Clin. Exp. Pathol. 2013, 6, 650.
- Fasoulakis, Z.; Koutras, A.; Antsaklis, P.; Theodora, M.; Valsamaki, A.; Daskalakis, G.; Kontomanolis, E.N. Intrauterine Growth Restriction Due to Gestational Diabetes: From Pathophysiology to Diagnosis and Management. Medicina 2023, 59, 1139.
- Marchi, J.; Berg, M.; Dencker, A.; Olander, E.K.; Begley, C. Risks Associated with Obesity in Pregnancy, for the Mother and Baby: A Systematic Review of Reviews. Obes. Rev. 2015, 16, 621–638.
- Johns, E.C.; Denison, F.C.; Norman, J.E.; Reynolds, R.M. Gestational Diabetes Mellitus: Mechanisms, Treatment, and Complications. Trends Endocrinol. Metab. 2018, 29, 743–754.
- Mao, J.; Zheng, Q.; Jin, L. Physiological Function of the Dynamic Oxygen Signaling Pathway at the Maternal-Fetal Interface. J. Reprod. Immunol. 2022, 151, 103626.
- Desoye, G.; Carter, A.M. Fetoplacental Oxygen Homeostasis in Pregnancies with Maternal Diabetes Mellitus and Obesity. Nat. Rev. Endocrinol. 2022, 18, 593–607.
- Torres-Cuevas, I.; Parra-Llorca, A.; Sánchez-Illana, A.; Nuñez-Ramiro, A.; Kuligowski, J.; Cháfer-Pericás, C.; Cernada, M.; Escobar, J.; Vento, M. Oxygen and Oxidative Stress in the Perinatal Period. Redox Biol. 2017, 12, 674–681.
- Mutinati, M.; Piccinno, M.; Roncetti, M.; Campanile, D.; Rizzo, A.; Sciorsci, R. Oxidative Stress during Pregnancy in the Sheep. Reprod. Domest. Anim. 2013, 48, 353–357.
- Al-Gubory, K.H.; Fowler, P.A.; Garrel, C. The Roles of Cellular Reactive Oxygen Species, Oxidative Stress and Antioxidants in Pregnancy Outcomes. Int. J. Biochem. Cell Biol. 2010, 42, 1634–1650.
- Hussain, T.; Murtaza, G.; Metwally, E.; Kalhoro, D.H.; Kalhoro, M.S.; Rahu, B.A.; Sahito, R.G.A.; Yin, Y.; Yang, H.; Chughtai, M.I.; et al. The Role of Oxidative Stress and Antioxidant Balance in Pregnancy. Mediat. Inflamm. 2021, 2021, 9962860.
- Sobrevia, L.; Valero, P.; Grismaldo, A.; Villalobos-Labra, R.; Pardo, F.; Subiabre, M.; Armstrong, G.; Toledo, F.; Vega, S.; Cornejo, M.; et al. Mitochondrial Dysfunction in the Fetoplacental Unit in Gestational Diabetes Mellitus. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2020, 1866, 165948.
- Gyllenhammer, L.E.; Entringer, S.; Buss, C.; Wadhwa, P.D. Developmental Programming of Mitochondrial Biology: A Conceptual Framework and Review. Proc. R. Soc. B Biol. Sci. 2020, 287, 20192713.
- Holland, O.; Dekker Nitert, M.; Gallo, L.A.; Vejzovic, M.; Fisher, J.J.; Perkins, A.V. Review: Placental Mitochondrial Function and Structure in Gestational Disorders. Placenta 2017, 54, 2–9.
- Meng, Q.; Shao, L.; Luo, X.; Mu, Y.; Xu, W.; Gao, C.; Gao, L.; Liu, J.; Cui, Y. Ultrastructure of Placenta of Gravidas with Gestational Diabetes Mellitus. Obstet. Gynecol. Int. 2015, 2015, 283124.
- Protasoni, M.; Zeviani, M. Mitochondrial Structure and Bioenergetics in Normal and Disease Conditions. Int. J. Mol. Sci. 2021, 22, 586.
- Fisher, J.J.; Vanderpeet, C.L.; Bartho, L.A.; McKeating, D.R.; Cuffe, J.S.M.; Holland, O.J.; Perkins, A.V. Mitochondrial Dysfunction in Placental Trophoblast Cells Experiencing Gestational Diabetes Mellitus. J. Physiol. 2021, 599, 1291–1305.
- Valent, A.M.; Choi, H.; Kolahi, K.S.; Thornburg, K.L. Hyperglycemia and Gestational Diabetes Suppress Placental Glycolysis and Mitochondrial Function and Alter Lipid Processing. FASEB J. 2021, 35, e21423.
- Jiang, S.; Teague, A.M.; Tryggestad, J.B.; Aston, C.E.; Lyons, T.; Chernausek, S.D. Effects of Maternal Diabetes and Fetal Sex on Human Placenta Mitochondrial Biogenesis. Placenta 2017, 57, 26–32.
- Abbade, J.; Klemetti, M.M.; Farrell, A.; Ermini, L.; Gillmore, T.; Sallais, J.; Tagliaferro, A.; Post, M.; Caniggia, I. Increased Placental Mitochondrial Fusion in Gestational Diabetes Mellitus: An Adaptive Mechanism to Optimize Feto-Placental Metabolic Homeostasis? BMJ Open Diabetes Res. Care 2020, 8, e000923.
- Kolac, U.K.; Kurek Eken, M.; Ünübol, M.; Donmez Yalcin, G.; Yalcin, A. The Effect of Gestational Diabetes on the Expression of Mitochondrial Fusion Proteins in Placental Tissue. Placenta 2021, 115, 106–114.
- Hori, A.; Yoshida, M.; Ling, F. Mitochondrial Fusion Increases the Mitochondrial DNA Copy Number in Budding Yeast. Genes Cells 2011, 16, 527–544.
- McElwain, C.; McCarthy, C.M. Investigating Mitochondrial Dysfunction in Gestational Diabetes Mellitus and Elucidating If BMI Is a Causative Mediator. Eur. J. Obstet. Gynecol. Reprod. Biol. 2020, 251, 60–65.
- Crovetto, F.; Lattuada, D.; Rossi, G.; Mangano, S.; Somigliana, E.; Bolis, G.; Fedele, L. A Role for Mitochondria in Gestational Diabetes Mellitus? Gynecol. Endocrinol. 2013, 29, 259–262.
- Qiu, C.; Hevner, K.; Abetew, D.; Sedensky, M.; Morgan, P.; Enquobahrie, D.; Williams, M. Mitochondrial DNA Copy Number and Oxidative DNA Damage in Placental Tissues from Gestational Diabetes and Control Pregnancies: A Pilot Study. Clin. Lab. 2013, 59, 655–660.
- Chen, X.; Scholl, T.O. Oxidative Stress: Changes in Pregnancy and with Gestational Diabetes Mellitus. Curr. Diab. Rep. 2005, 5, 282–288.
- Biri, A.; Onan, A.; Devrim, E.; Babacan, F.; Kavutcu, M.; Durak, İ. Oxidant Status in Maternal and Cord Plasma and Placental Tissue in Gestational Diabetes. Placenta 2006, 27, 327–332.
- Diceglie, C.; Anelli, G.M.; Martelli, C.; Serati, A.; Lo Dico, A.; Lisso, F.; Parisi, F.; Novielli, C.; Paleari, R.; Cetin, I.; et al. Placental Antioxidant Defenses and Autophagy-Related Genes in Maternal Obesity and Gestational Diabetes Mellitus. Nutrients 2021, 13, 1303.
- Kim, J.; Piao, Y.; Pak, Y.K.; Chung, D.; Han, Y.M.; Hong, J.S.; Jun, E.J.; Shim, J.-Y.; Choi, J.; Kim, C.J. Umbilical Cord Mesenchymal Stromal Cells Affected by Gestational Diabetes Mellitus Display Premature Aging and Mitochondrial Dysfunction. Stem Cells Dev. 2015, 24, 575–586.
- Almalki, S.G.; Agrawal, D.K. Key Transcription Factors in the Differentiation of Mesenchymal Stem Cells. Differentiation 2016, 92, 41–51.
- Cornejo, M.; Fuentes, G.; Valero, P.; Vega, S.; Grismaldo, A.; Toledo, F.; Pardo, F.; Moore-Carrasco, R.; Subiabre, M.; Casanello, P.; et al. Gestational Diabesity and Foetoplacental Vascular Dysfunction. Acta Physiol. 2021, 232, e13671.
- Hu, C.; Yan, Y.; Ji, F.; Zhou, H. Maternal Obesity Increases Oxidative Stress in Placenta and It Is Associated With Intestinal Microbiota. Front. Cell. Infect. Microbiol. 2021, 11, 671347.
- Mele, J.; Muralimanoharan, S.; Maloyan, A.; Myatt, L. Impaired Mitochondrial Function in Human Placenta with Increased Maternal Adiposity. Am. J. Physiol.-Endocrinol. Metab. 2014, 307, E419–E425.
- Hu, C.; Yang, Y.; Li, J.; Wang, H.; Cheng, C.; Yang, L.; Li, Q.; Deng, J.; Liang, Z.; Yin, Y.; et al. Maternal Diet-Induced Obesity Compromises Oxidative Stress Status and Angiogenesis in the Porcine Placenta by Upregulating Nox2 Expression. Oxid. Med. Cell. Longev. 2019, 2019, 2481592.
- Ballesteros-Guzmán, A.K.; Carrasco-Legleu, C.E.; Levario-Carrillo, M.; Chávez-Corral, D.V.; Sánchez-Ramírez, B.; Mariñelarena-Carrillo, E.O.; Guerrero-Salgado, F.; Reza-López, S.A. Prepregnancy Obesity, Maternal Dietary Intake, and Oxidative Stress Biomarkers in the Fetomaternal Unit. Biomed. Res. Int. 2019, 2019, 5070453.
- Mandò, C.; De Palma, C.; Stampalija, T.; Anelli, G.M.; Figus, M.; Novielli, C.; Parisi, F.; Clementi, E.; Ferrazzi, E.; Cetin, I. Placental Mitochondrial Content and Function in Intrauterine Growth Restriction and Preeclampsia. Am. J. Physiol.-Endocrinol. Metab. 2014, 306, E404–E413.
- Chiaratti, M.R.; Malik, S.; Diot, A.; Rapa, E.; Macleod, L.; Morten, K.; Vatish, M.; Boyd, R.; Poulton, J. Is Placental Mitochondrial Function a Regulator That Matches Fetal and Placental Growth to Maternal Nutrient Intake in the Mouse? PLoS ONE 2015, 10, e0130631.
- Aljunaidy, M.M.; Morton, J.S.; Kirschenman, R.; Phillips, T.; Case, C.P.; Cooke, C.-L.M.; Davidge, S.T. Maternal Treatment with a Placental-Targeted Antioxidant (MitoQ) Impacts Offspring Cardiovascular Function in a Rat Model of Prenatal Hypoxia. Pharmacol. Res. 2018, 134, 332–342.
- Richter, H.G.; Camm, E.J.; Modi, B.N.; Naeem, F.; Cross, C.M.; Cindrova-Davies, T.; Spasic-Boskovic, O.; Dunster, C.; Mudway, I.S.; Kelly, F.J.; et al. Ascorbate Prevents Placental Oxidative Stress and Enhances Birth Weight in Hypoxic Pregnancy in Rats. J. Physiol. 2012, 590, 1377–1387.
- Keenaghan, M.; Sun, L.; Wang, A.; Hyodo, E.; Homma, S.; Ten, V.S. Intrauterine Growth Restriction Impairs Right Ventricular Response to Hypoxia in Adult Male Rats. Pediatr. Res. 2016, 80, 547–553.
- Pereira, S.P.; Tavares, L.C.; Duarte, A.I.; Baldeiras, I.; Cunha-Oliveira, T.; Martins, J.D.; Santos, M.S.; Maloyan, A.; Moreno, A.J.; Cox, L.A.; et al. Sex-Dependent Vulnerability of Fetal Nonhuman Primate Cardiac Mitochondria to Moderate Maternal Nutrient Reduction. Clin. Sci. 2021, 135, 1103–1126.
- Guitart-Mampel, M.; Gonzalez-Tendero, A.; Niñerola, S.; Morén, C.; Catalán-Garcia, M.; González-Casacuberta, I.; Juárez-Flores, D.L.; Ugarteburu, O.; Matalonga, L.; Cascajo, M.V.; et al. Cardiac and Placental Mitochondrial Characterization in a Rabbit Model of Intrauterine Growth Restriction. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2018, 1862, 1157–1167.
- Peterside, I.E.; Selak, M.A.; Simmons, R.A. Impaired Oxidative Phosphorylation in Hepatic Mitochondria in Growth-Retarded Rats. Am. J. Physiol.-Endocrinol. Metab. 2003, 285, E1258–E1266.
- Rains, M.E.; Muncie, C.B.; Pang, Y.; Fan, L.-W.; Tien, L.-T.; Ojeda, N.B. Oxidative Stress and Neurodevelopmental Outcomes in Rat Offspring with Intrauterine Growth Restriction Induced by Reduced Uterine Perfusion. Brain Sci. 2021, 11, 78.
- Devarajan, A.; Rajasekaran, N.S.; Valburg, C.; Ganapathy, E.; Bindra, S.; Freije, W.A. Maternal Perinatal Calorie Restriction Temporally Regulates the Hepatic Autophagy and Redox Status in Male Rat. Free Radic. Biol. Med. 2019, 130, 592–600.
- Grilo, L.F.; Diniz, M.S.; Tocantins, C.; Areia, A.L.; Pereira, S.P. The Endocrine–Metabolic Axis Regulation in Offspring Exposed to Maternal Obesity—Cause or Consequence in Metabolic Disease Programming? Obesities 2022, 2, 236–255.
- Grilo, L.F.; Martins, J.D.; Diniz, M.S.; Tocantins, C.; Cavallaro, C.H.; Baldeiras, I.; Cunha-Oliveira, T.; Ford, S.; Nathanielsz, P.W.; Oliveira, P.J.; et al. Maternal Hepatic Adaptations during Obese Pregnancy Encompass Lobe-Specific Mitochondrial Alterations and Oxidative Stress. Clin. Sci. 2023, 137, 1347–1372.
- Mirończuk-Chodakowska, I.; Witkowska, A.M.; Zujko, M.E. Endogenous Non-Enzymatic Antioxidants in the Human Body. Adv. Med. Sci. 2018, 63, 68–78.
- Jena, A.B.; Samal, R.R.; Bhol, N.K.; Duttaroy, A.K. Cellular Red-Ox System in Health and Disease: The Latest Update. Biomed. Pharmacother. 2023, 162, 114606.
- Pisoschi, A.M.; Pop, A. The Role of Antioxidants in the Chemistry of Oxidative Stress: A Review. Eur. J. Med. Chem. 2015, 97, 55–74.
- Bentinger, M.; Brismar, K.; Dallner, G. The Antioxidant Role of Coenzyme Q. Mitochondrion 2007, 7, S41–S50.
- Teixeira, J.; Chavarria, D.; Borges, F.; Wojtczak, L.; Wieckowski, M.R.; Karkucinska-Wieckowska, A.; Oliveira, P.J. Dietary Polyphenols and Mitochondrial Function: Role in Health and Disease. Curr. Med. Chem. 2019, 26, 3376–3406.
- Rajendran, P.; Abdelsalam, S.A.; Renu, K.; Veeraraghavan, V.; Ben Ammar, R.; Ahmed, E.A. Polyphenols as Potent Epigenetics Agents for Cancer. Int. J. Mol. Sci. 2022, 23, 11712.
- Croft, K.D. Dietary Polyphenols: Antioxidants or Not? Arch. Biochem. Biophys. 2016, 595, 120–124.
- Nani, A.; Murtaza, B.; Sayed Khan, A.; Khan, N.A.; Hichami, A. Antioxidant and Anti-Inflammatory Potential of Polyphenols Contained in Mediterranean Diet in Obesity: Molecular Mechanisms. Molecules 2021, 26, 985.
- Jantan, I.; Haque, M.A.; Arshad, L.; Harikrishnan, H.; Septama, A.W.; Mohamed-Hussein, Z.-A. Dietary Polyphenols Suppress Chronic Inflammation by Modulation of Multiple Inflammation-Associated Cell Signaling Pathways. J. Nutr. Biochem. 2021, 93, 108634.
- Shahwan, M.; Alhumaydhi, F.; Ashraf, G.M.; Hasan, P.M.Z.; Shamsi, A. Role of Polyphenols in Combating Type 2 Diabetes and Insulin Resistance. Int. J. Biol. Macromol. 2022, 206, 567–579.
- Park, S.-J.; Ahmad, F.; Philp, A.; Baar, K.; Williams, T.; Luo, H.; Ke, H.; Rehmann, H.; Taussig, R.; Brown, A.L.; et al. Resveratrol Ameliorates Aging-Related Metabolic Phenotypes by Inhibiting CAMP Phosphodiesterases. Cell 2012, 148, 421–433.
- Lagouge, M.; Argmann, C.; Gerhart-Hines, Z.; Meziane, H.; Lerin, C.; Daussin, F.; Messadeq, N.; Milne, J.; Lambert, P.; Elliott, P.; et al. Resveratrol Improves Mitochondrial Function and Protects against Metabolic Disease by Activating SIRT1 and PGC-1α. Cell 2006, 127, 1109–1122.
- Hoseini, A.; Namazi, G.; Farrokhian, A.; Reiner, Ž.; Aghadavod, E.; Bahmani, F.; Asemi, Z. The Effects of Resveratrol on Metabolic Status in Patients with Type 2 Diabetes Mellitus and Coronary Heart Disease. Food Funct. 2019, 10, 6042–6051.
- Cao, M.; Lu, X.; Liu, G.; Su, Y.; Li, Y.; Zhou, J. Resveratrol Attenuates Type 2 Diabetes Mellitus by Mediating Mitochondrial Biogenesis and Lipid Metabolism via Sirtuin Type 1. Exp. Ther. Med. 2018, 15, 576–584.
- Ren, B.; Zhang, Y.; Liu, S.; Cheng, X.; Yang, X.; Cui, X.; Zhao, X.; Zhao, H.; Hao, M.; Li, M.; et al. Curcumin Alleviates Oxidative Stress and Inhibits Apoptosis in Diabetic Cardiomyopathy via Sirt1-Foxo1 and PI3K-Akt Signalling Pathways. J. Cell. Mol. Med. 2020, 24, 12355–12367.
- Hamidie, R.D.R.; Shibaguchi, T.; Yamada, T.; Koma, R.; Ishizawa, R.; Saito, Y.; Hosoi, T.; Masuda, K. Curcumin Induces Mitochondrial Biogenesis by Increasing Cyclic AMP Levels via Phosphodiesterase 4A Inhibition in Skeletal Muscle. Br. J. Nutr. 2021, 126, 1642–1650.
- Li, Y.; Luo, W.; Cheng, X.; Xiang, H.; He, B.; Zhang, Q.; Peng, W. Curcumin Attenuates Isoniazid-induced Hepatotoxicity by Upregulating the SIRT1/PGC-1α/NRF1 Pathway. J. Appl. Toxicol. 2022, 42, 1192–1204.
- Ren, T.; Huang, C.; Cheng, M. Dietary Blueberry and Bifidobacteria Attenuate Nonalcoholic Fatty Liver Disease in Rats by Affecting SIRT1-Mediated Signaling Pathway. Oxid. Med. Cell. Longev. 2014, 2014, 469059.
- Jiménez-Flores, L.; López-Briones, S.; Macías-Cervantes, M.; Ramírez-Emiliano, J.; Pérez-Vázquez, V. A PPARγ, NF-ΚB and AMPK-Dependent Mechanism May Be Involved in the Beneficial Effects of Curcumin in the Diabetic Db/Db Mice Liver. Molecules 2014, 19, 8289–8302.
- Zendedel, E.; Butler, A.E.; Atkin, S.L.; Sahebkar, A. Impact of Curcumin on Sirtuins: A Review. J. Cell Biochem. 2018, 119, 10291–10300.
- Domazetovic, V.; Marcucci, G.; Pierucci, F.; Bruno, G.; Di Cesare Mannelli, L.; Ghelardini, C.; Brandi, M.L.; Iantomasi, T.; Meacci, E.; Vincenzini, M.T. Blueberry Juice Protects Osteocytes and Bone Precursor Cells against Oxidative Stress Partly through SIRT1. FEBS Open Bio 2019, 9, 1082–1096.
- Ungvari, Z.; Bagi, Z.; Feher, A.; Recchia, F.A.; Sonntag, W.E.; Pearson, K.; de Cabo, R.; Csiszar, A. Resveratrol Confers Endothelial Protection via Activation of the Antioxidant Transcription Factor Nrf2. Am. J. Physiol.-Heart Circ. Physiol. 2010, 299, H18–H24.
- Wei, Z.; Shaohuan, Q.; Pinfang, K.; Chao, S. Curcumin Attenuates Ferroptosis-Induced Myocardial Injury in Diabetic Cardiomyopathy through the Nrf2 Pathway. Cardiovasc. Ther. 2022, 2022, 3159717.
- Lin, X.; Bai, D.; Wei, Z.; Zhang, Y.; Huang, Y.; Deng, H.; Huang, X. Curcumin Attenuates Oxidative Stress in RAW264.7 Cells by Increasing the Activity of Antioxidant Enzymes and Activating the Nrf2-Keap1 Pathway. PLoS ONE 2019, 14, e0216711.
- Ashrafizadeh, M.; Ahmadi, Z.; Mohammadinejad, R.; Farkhondeh, T.; Samarghandian, S. Curcumin Activates the Nrf2 Pathway and Induces Cellular Protection Against Oxidative Injury. Curr. Mol. Med. 2020, 20, 116–133.
- Li, C.; Tan, Y.; Wu, J.; Ma, Q.; Bai, S.; Xia, Z.; Wan, X.; Liang, J. Resveratrol Improves Bnip3-Related Mitophagy and Attenuates High-Fat-Induced Endothelial Dysfunction. Front. Cell. Dev. Biol. 2020, 8, 00796.
- Brenjian, S.; Moini, A.; Yamini, N.; Kashani, L.; Faridmojtahedi, M.; Bahramrezaie, M.; Khodarahmian, M.; Amidi, F. Resveratrol Treatment in Patients with Polycystic Ovary Syndrome Decreased Pro-inflammatory and Endoplasmic Reticulum Stress Markers. Am. J. Reprod. Immunol. 2020, 83, e13186.
- Csiszar, A.; Smith, K.; Labinskyy, N.; Orosz, Z.; Rivera, A.; Ungvari, Z. Resveratrol Attenuates TNF-α-Induced Activation of Coronary Arterial Endothelial Cells: Role of NF-ΚB Inhibition. Am. J. Physiol.-Heart Circ. Physiol. 2006, 291, H1694–H1699.
- Wang, Q.; Ye, C.; Sun, S.; Li, R.; Shi, X.; Wang, S.; Zeng, X.; Kuang, N.; Liu, Y.; Shi, Q.; et al. Curcumin Attenuates Collagen-Induced Rat Arthritis via Anti-Inflammatory and Apoptotic Effects. Int. Immunopharmacol. 2019, 72, 292–300.
- Aldini, G.; Altomare, A.; Baron, G.; Vistoli, G.; Carini, M.; Borsani, L.; Sergio, F. N-Acetylcysteine as an Antioxidant and Disulphide Breaking Agent: The Reasons Why. Free Radic. Res. 2018, 52, 751–762.
- Jin, H.; Kanthasamy, A.; Ghosh, A.; Anantharam, V.; Kalyanaraman, B.; Kanthasamy, A.G. Mitochondria-Targeted Antioxidants for Treatment of Parkinson’s Disease: Preclinical and Clinical Outcomes. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2014, 1842, 1282–1294.
- Rushworth, G.F.; Megson, I.L. Existing and Potential Therapeutic Uses for N-Acetylcysteine: The Need for Conversion to Intracellular Glutathione for Antioxidant Benefits. Pharmacol. Ther. 2014, 141, 150–159.
- Raghu, G.; Berk, M.; Campochiaro, P.A.; Jaeschke, H.; Marenzi, G.; Richeldi, L.; Wen, F.-Q.; Nicoletti, F.; Calverley, P.M.A. The Multifaceted Therapeutic Role of N-Acetylcysteine (NAC) in Disorders Characterized by Oxidative Stress. Curr. Neuropharmacol. 2021, 19, 1202–1224.
- Barrozo, L.G.; Paulino, L.R.F.M.; Silva, B.R.; Barbalho, E.C.; Nascimento, D.R.; Neto, M.F.L.; Silva, J.R.V. N-Acetyl-Cysteine and the Control of Oxidative Stress during in Vitro Ovarian Follicle Growth, Oocyte Maturation, Embryo Development and Cryopreservation. Anim. Reprod. Sci. 2021, 231, 106801.
- Wang, S.; Liu, G.; Jia, T.; Wang, C.; Lu, X.; Tian, L.; Yang, Q.; Zhu, C. Protection Against Post-Resuscitation Acute Kidney Injury by N-Acetylcysteine via Activation of the Nrf2/HO-1 Pathway. Front. Med. 2022, 9, 848491.
- Teixeira, J.; Deus, C.M.; Borges, F.; Oliveira, P.J. Mitochondria: Targeting Mitochondrial Reactive Oxygen Species with Mitochondriotropic Polyphenolic-Based Antioxidants. Int. J. Biochem. Cell Biol. 2018, 97, 98–103.
- Marshall, N.E.; Abrams, B.; Barbour, L.A.; Catalano, P.; Christian, P.; Friedman, J.E.; Hay, W.W.; Hernandez, T.L.; Krebs, N.F.; Oken, E.; et al. The Importance of Nutrition in Pregnancy and Lactation: Lifelong Consequences. Am. J. Obstet. Gynecol. 2022, 226, 607–632.
- Ota, E.; Tobe-Gai, R.; Mori, R.; Farrar, D. Antenatal Dietary Advice and Supplementation to Increase Energy and Protein Intake. In Cochrane Database of Systematic Reviews; Ota, E., Ed.; John Wiley & Sons, Ltd.: Chichester, UK, 2012.
- Weckman, A.M.; McDonald, C.R.; Baxter, J.-A.B.; Fawzi, W.W.; Conroy, A.L.; Kain, K.C. Perspective: L-Arginine and L-Citrulline Supplementation in Pregnancy: A Potential Strategy to Improve Birth Outcomes in Low-Resource Settings. Adv. Nutr. 2019, 10, 765–777.
- Liang, M.; Wang, Z.; Li, H.; Cai, L.; Pan, J.; He, H.; Wu, Q.; Tang, Y.; Ma, J.; Yang, L. L-Arginine Induces Antioxidant Response to Prevent Oxidative Stress via Stimulation of Glutathione Synthesis and Activation of Nrf2 Pathway. Food Chem. Toxicol. 2018, 115, 315–328.
- Imdad, A.; Bhutta, Z.A. Routine Iron/Folate Supplementation during Pregnancy: Effect on Maternal Anaemia and Birth Outcomes. Paediatr. Perinat. Epidemiol. 2012, 26, 168–177.
- Kurtoglu, E.; Ugur, A.; Baltaci, A.K.; Undar, L. Effect of Iron Supplementation on Oxidative Stress and Antioxidant Status in Iron-Deficiency Anemia. Biol. Trace Elem. Res. 2003, 96, 117–123.
- Georgieff, M.K.; Krebs, N.F.; Cusick, S.E. The Benefits and Risks of Iron Supplementation in Pregnancy and Childhood. Annu. Rev. Nutr. 2019, 39, 121–146.
More