Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by George Briassoulis and Version 2 by Peter Tang.
Zinc is a structural component of proteins, functions as a catalytic co-factor in DNA synthesis and transcription of hundreds of enzymes, and has a regulatory role in protein–DNA interactions of zinc-finger proteins.
- micronutrient
- zinc
- bioavailability
- antioxidant
- anti-inflammatory
1. Introduction
Zinc is an essential micronutrient needed to ensure the proper functioning of crucial biochemical and metabolic processes. The role of zinc in the body can be grouped into three general functional classes: (a) structural component in proteins; (b) catalytic co-factor in DNA synthesis and RNA transcription of hundreds of enzymes such as polymerases, carboxy peptidases, superoxide dismutase, and carbonic anhydrase [1][9]; and (c) regulatory in protein–DNA interactions of zinc-finger proteins, including many transcription factors [2][10], inhibitory of viral replication [3][11], and modulatory of inflammation [4][12] and oxidative stress [5][13]. Among them, the most extensively studied regulatory functions are antioxidant activity, the effect on inflammation, immune regulations, and regulated cell death.
2. Antioxidant Activity
Although the production of reactive oxygen species (ROS) aims to eradicate pathogens, excessive oxidative stress in sepsis leads to cell membrane lipid peroxidation, protein oxidation, mitochondrial damage, inflammation, and increased inducible nitric oxide synthetase (iNOS)-induced NO, apoptosis and necroptosis [6][14]. The main antioxidant proteins, including superoxide dismutase (SOD), glutathione, catalase, thioredoxin, and peroxiredoxin, regulate the redox balance in the mitochondria [7][15]. In a clinical study, elderly individuals who received zinc supplementation exhibited notably reduced rates of infections and lower levels of plasma oxidative stress markers when compared to those in the placebo group [8][16]. Hence, zinc could be of potential benefit as antioxidant therapy for redox balance restoration in sepsis and COVID-19 [9][17].
Zinc acts as an antioxidant by neutralizing free radicals directly by glutathione, affecting the expression of the rate-limiting enzyme of de novo synthesis glutathione, glutamate-cysteine ligase [10][18]. Zinc enhances the effectiveness of Cu/Zn SOD by serving as a structural component. This enzyme transforms superoxide (O2−) into the less harmful hydrogen peroxide (H2O2) and oxygen (O2), thereby reducing the harmful effects of reactive oxygen species (ROS) [11][19]. Metallothioneins are involved in the reduction of hydroxyl radicals, acting as cytoprotective agents and electrophilic scavengers, maintaining zinc-related cell homeostasis [12][20].
In murine sepsis, zinc was shown to activate the nuclear factor-erythroid 2-related factor 2 (Nrf2) transcription factor, which regulates glutathione peroxidase 2 (GPx2), glutamate-cysteine ligase catalytic subunit (GCLC), thioredoxin reductase 1, members of the glutathione-S-transferase (GST), heme-oxygenase-1 (HO-1), and SOD by binding to the anti-oxidant responsive cis-acting enhancer sequence elements [13][21]. Also, different forms of zinc supplementation (zinc methionine, zinc sulphate, and nano zinc oxide) improved serum and seminal plasma SOD and catalase, along with physiological parameters, of heat-stressed male rabbits [14][22]. Zinc was shown to increase metallothionein in the liver of endotoxin/ Zn2+-adequate diet rats, inhibit endotoxin lethality in Zn2+-treated animals, and protect against endotoxin-associated mediators [15][23].
The indirect antioxidant function of zinc is to compete with redox-active metals for negative charges in the lipid bilayer [16][24], protecting the cell membrane from the lipid oxidation [17][25], and interacting with thiol and sulfhydryl groups, preventing intramolecular disulfide formation [18][26]. A second mechanism consists of antagonizing metal-catalysed transition reactions [12][20] by exchanging redox-active metals, such as copper and iron, in certain binding sites attenuating cellular site-specific oxidative injury in the setting of severe infection. It is part of the antioxidant defence to stabilize cytosolic Zn/Cu superoxide dismutase, inhibiting the pro-oxidant enzyme nicotinamide adenine dinucleotide phosphate oxidase (NADPH)-Oxidase producing superoxide radicals (O2−) and inducible nitric oxide synthase (iNOS) [19][27], and inducing the expression of cysteine-rich metallothioneins [20][28].
Experimentally, anti-oxidant genes were noticeably improved with a dietary zinc nanoparticles diet and iNOS, heat shock protein-70, and DNA damage-inducible protein, were significantly upregulated in fish [21][29] and mice [22][30]. In rats, zinc oxide nanoparticles demonstrated antioxidant properties by effectively reducing serum cyclooxygenase-2 (COX-2) enzyme activity, decreasing tissue nuclear factor—kappa B (NF-κB) levels, and lowering blood hypoxia-inducible factors (HIF-1α) levels [23][31]. Supplementation of zinc-enriched probiotics of Wistar rats significantly enhanced glutathione content, glutathione-peroxidase and superoxide-dismutase activity, and decreased malondialdehyde content while the expression of glutathione peroxidase 1 (GPx1) and SOD1 genes significantly increased [24][32]. Also, exposure of magur catfish to zinc oxide nanoparticles, stimulated the NO production by hepatocytes because of induction of iNOS activity, higher expression of nos2 gene and iNOS protein [25][33].
3. Effect on Inflammation
Various pro-inflammatory transcription factors, such as NF-κB and activator protein-1 (AP-1), have the capability to trigger the generation of ROS, leading to the secretion of inflammatory cytokines. This, in turn, exacerbates oxidative stress [26][34]. Cellular transcriptional response to ROS is mediated mainly by activation of mitogen-activated protein (MAP) kinases, cysteines Cys65 and Cys93 in redox effector factor (Ref-1), the redox-dependent modification of transcription factors p53, activating transcription factor/cAMP-response, element–binding protein (ATF/CREB), hypoxia-inducible factor (HIF)-1α, and HIF-like factor [27][28][35,36]. Superoxide anions undergo oxidative phosphorylation with the oxidative systems consisting of xanthine oxidase, nicotinamide adenine dinucleotide phosphate (NADPH) oxidases, 5-lipoxygenase, and cyclooxygenase [29][37]. Strong experimental evidence shows that the proteasome partially processes phosphorylation of p105 and p100 precursor NF-κB proteins at S893 and S907 residues to produce NF-κB subunits p50 and p52. The p105 and p100 precursor proteins contain C-terminal ankyrin repeats conferring IκB-like function by sequestering interacting NF-κB subunits in the cytoplasm. The p50 NF-κB subunit is phosphorylated by protein kinase A (PKA) at S337, by checkpoint kinase 1 (Chk1) at 328 triggered by DNA damage, and by DNA-dependent protein kinase (DNA-PK) responded to TNF-α stimulus at S20 [30][38].
NF-κB is phosphorylated downstream in responding to toll-like receptors (TLRs), antigens, cytokines, growth factor receptors, oxidative stimuli, and viral products. In the cytoplasm, the phosphorylation of NF-κB at specific NH2-terminal serine residues degrades the inhibitory subunit inhibitors of κB (IκB), activating the IκB kinase (IKK) complex of IKKα and IKKβ. Phosphorylation of IκBα by the IKK complex triggers its recognition by beta-transducing repeats-containing proteins (β-TrCP), substrate recognition subunits for the SCFβ-TrCP E3 ubiquitin ligases, leading to polyubiquitination and proteasome degradation. The released NF-κB dimers then translocate to the nucleus, where they recognise and bind specific DNA sequences, termed κB sites, activating the transcription of many different target-proinflammatory genes [31][39]. The NF-κB signalling pathway regulates inflammatory responses through the IκB kinase (IKK) complex to promote cell survival. The activation of IKKα and IKKβ, induced by TNF stimulation and involving the ubiquitylation of receptor-interacting serine/threonine-protein kinase-1 (RIPK1) and NF-κB essential modifier (NEMO), plays a pivotal role in promoting cell survival through the NF-κB signalling pathway [32][40]. TNF receptor 1 (TNFR1) ligation activates the NF-κB signaling pathway acting as a cell death determinant because NF-κB transcription factors induce the expression of anti-apoptotic genes. Also, in the TGFβ-activated kinase-1 (TAK1) complex, TAB2 contains Lys63-linked ubiquitin-binding domains, interacts with RIPK1 and NEMO [33][41], and activates NF-κB signalling by initiating the IKK complex [34][42]. NF-κB activation increases the expression of the adhesion molecules E-selectin, VCAM-1, and intercellular adhesion molecule 1 (ICAM-1) and mediates the synthesis of cytokines and cyclooxygenase 2 (Cox-2) [31][39]. NF-κB has also been shown to function in concert with other transcription factors, such as AP-1 [35][43].
Upon zinc supplementation, zinc receptor metal regulatory transcription factor 1 (MTF-1) and other transcription factors responsible for cellular adaptation to oxidative stress enhance gene expression within the nucleus. This enhancement occurs by increasing DNA binding and recruiting various co-activators, thereby maintaining cellular homeostasis [36][44]. The release of zinc from metallothioneins activates NF-κB and influences its ability to transmit pro-inflammatory signals to the nucleus, regulating the expression of DNA for transcription factors involved in the inflammation process. Inflammatory states have shown a coordinated interplay among zinc, metallothioneins, MTF-1, and pro-inflammatory cytokines like IL-6 and TNF-α, orchestrated by the transcription factor NF-κB [36][44]. Zinc can attenuate the pro-inflammatory response through cell uptake by the zinc transporter protein ZIP14, also known as SLC39A14 (Solute Carrier Family 39 Member 14), which plays a crucial role in zinc homeostasis within the body. ZIP14 facilitates the movement of zinc from storage sites (liver) to immune cells at the site of infection in sepsis, by facilitating the redistribution of zinc to immune cells, supporting immune function, modulating inflammation, and aiding in antioxidant defence [37][45]. Current evidence indicates that zinc modulates NF-κB signalling at various levels, primarily by regulating the activity of proteins and enzymes involved in the NF-κB pathway. Some ways zinc can influence NF-κB signalling are the inhibition of IKK activation, enhancement of IκB stability, modulation of redox status, regulation of zinc finger proteins, influence on immune cell function, and epigenetic regulation [38][46]. Zinc can inhibit the activation of IKK, a kinase that phosphorylates IκB (inhibitor of NF-κB), leading to its degradation. This prevents the release and translocation of NF-κB to the nucleus [39][47]. Simultaneously, zinc enhances the stability of IκB, which sequesters NF-κB in the cytoplasm. The activation of the NF-κB heterodimer, composed of p65-p50 and IκBα, occurs through the phosphorylation of IκB by the IκB kinase (IKK) complex. This leads to the ubiquitination and subsequent proteasomal degradation of IκB [40][48]. Zinc, through the inhibition of cyclic nucleotide phosphodiesterase (PDE), causes an increase in cyclic guanosine monophosphate (cGMP). This rise in cGMP activates protein kinase A (PKA) and, by phosphorylation, inhibits the protein kinase Raf-1. Moreover, zinc suppresses the activation of IKKβ induced by LPS [12][20]. Also, imported by the zinc transporter ZIP8, cytosolic zinc induces NF-κB inhibition downstream from mitogen-activated protein kinases (MAPKs) by blocking the IKK complex [41][49]. Finally, zinc negatively regulates the NF-κB pathway by inducing the expression of zinc-finger protein A20, which is the main negative regulator of the NF-κB activation [40][48]. Cytosolic zinc, transported via the zinc transporter ZIP8, induces the inhibition of NF-κB downstream of MAPKs by interfering with the IKK complex [39][47]. This, in turn, affects its target genes, such as TNF-α and IL-1-beta (IL-1β) [42][50], as well as chemoattractant proteins like monocyte chemoattractant protein-1 (MCP-1), and intercellular adhesion molecule-1 (ICAM-1) [43][51]. Additionally, zinc can indirectly modulate the NF-κB pathway by affecting the activity of immune cells that produce cytokines and other molecules involved in NF-κB signalling [44][52]. Furthermore, zinc can play a role in epigenetic regulation by influencing DNA methylation and histone modifications, potentially affecting the expression of genes involved in the NF-κB signalling pathway [45][53].
Zinc exerts an influence on the cellular redox status, mitigating oxidative stress and countering the activation of NF-κB signalling [46][54]. Given its importance for the function of various zinc finger proteins, some of which can interact with NF-κB, zinc can impact the activity and localization of NF-κB. Zinc fingers are autonomously folded domains structured around a zinc ion [47][55] with roles in DNA recognition, RNA packaging, transcriptional activation, protein folding, assembly, and regulation of necroptosis [48][56]. Research indicates that zinc plays a regulatory role in NF-κB transcription, mediated by the zinc-finger protein TNF-α-induced protein 3 (TNFAIP3) and the receptor signalling pathway activated by peroxisome proliferator-activated receptor (PPAR) [49][50][57,58]. TNFAIP3, also known as A20, is a widely expressed cytoplasmic signalling protein known for its anti-inflammatory, NF-κB inhibitory, and anti-necroptotic properties. A20 comprehensively regulates ubiquitin-dependent signals and modulates the duration and intensity of signalling by various proteins involved in the NF-κB pathway [49][51][52][57,59,60]. In TNF receptor (TNFR)- and toll-like receptor (TLR)-initiated pathways, TNFAIP3 serves as the primary negative regulator of NF-κB activation, impacting endothelial cell adhesion molecules and oxidative stress biomarkers [43][51]. Studies have shown that A20 functions as a negative regulator, maintaining the balance in the strength and duration of NF-κB activation by deubiquitinating receptor-interacting serine/threonine-protein kinase 1 (RIPK1) and TNF receptor-associated factor 2 (TRAF2), the components of the TNFR1 signalling complex (Figure 1). Moreover, A20’s deubiquitylase (DUB) activity restricts TRAF6-mediated and RIPK2-mediated activation of NF-κB during TLR/IL-1R and NOD signalling, respectively. In vitro, zinc upregulates the gene expression of A20 and peroxisome proliferator-activated receptor alpha (PPAR-α), both of which are zinc finger proteins with anti-inflammatory properties [12][20].
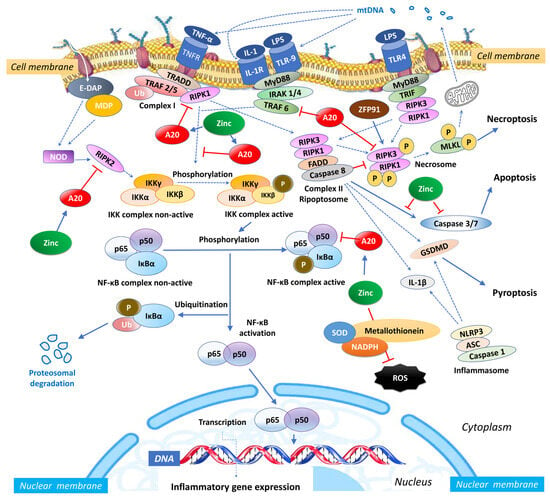
Figure 1. Intracellular zinc and A20 regulatory activities in sepsis. NF-κB: nuclear factor κB; TNF: tumour necrosis factor; TNFR: tumour necrosis factor receptor; TRAF: TNF receptor-associated factor; TRADD: death domain-containing adaptor protein; E-DAP: meso-diaminopimelic acid; MDP: muramyl dipeptide; RIPK: receptor-interacting serine/threonine-protein kinase; IKK: inhibitor of κB kinase; IκBα: nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor, alpha; Ub: ubiquitin; SOD: superoxide dismutase; NADPH: nicotinamide adenine dinucleotide phosphate; NLRP3: NLR Family Pyrin Domain Containing 3; ASC: apoptosis-associated speck-like protein containing a caspase recruitment domain; IL: interleukin; MLKL: mixed lineage kinase domain like pseudokinase; MyD88: myeloid differentiation primary response 88; IRAK-4: interleukin-1 receptor-associated kinase 4; ZFP91: zinc finger protein 91; LPS: Lipopolysaccharide; mtDNA: mitochondrial DNA; FADD: Fas-associated death domain; TLR: toll-like receptor; TRIF: TIR-domain-containing adaptor-inducing IFN-β; ROS: reactive oxygen species.
4. Immune Regulation
In sepsis, DAMPs, such as plasma mitochondrial DNA (mtDNA), serve as drivers of an immune or inflammatory response [53][61]. The DAMPs are released during sepsis-induced immune cell death, which is linked with multiple organ dysfunction [54][62]. Increasing evidence has shown that circulating mtDNA can trigger innate immunity by activating the Toll-like receptor 9/NF-κB pathway [55][63] or the NOD, LRR, and pyrin domain-containing protein 3 (NLRP3) inflammasome [56][64]. Zinc, in conjunction with various micronutrients, plays a role in DNA metabolism, resulting in reduced DNA damage and lowered levels of mtDNA in the bloodstream [57][65]. Conversely, lower zinc concentrations have been correlated with higher levels of mtDNA in patients with HIV and those coinfected with HIV and hepatitis C [58][66]. In an animal model, the presence of extracellular mtDNA activated the NLRP3 inflammasome, initiating inflammation via the Toll-like receptor 9 (TLR-9), mitogen-activated protein kinase (MAPK), and NF-κB pathways [59][67]. When monocytes were stimulated with mtDNA in vitro, it led to the production of TNF-α. Moreover, elevated levels of mtDNA in the plasma were correlated with increased in vivo levels of pro-inflammatory cytokines, including TNF-α, IL-6, Regulated upon Activation, Normal T Cell Expressed and Presumably Secreted (RANTES), and IL-1 receptor antagonist (IL-1ra) [60][68]. In both in vitro and in vivo settings, mtDNA activated neutrophils by promoting the phosphorylation of MAPK and inducing migration and degranulation of human polymorphonuclear neutrophils. This activation of neutrophils was mediated through TLR-9 and formyl peptide receptor-1 (FPR1) [61][69]. This relationship suggests that DAMPs might contribute to the persistent and dysregulated inflammatory response, leading to processes like apoptosis and necroptosis [54][62] and exacerbating organ dysfunction during sepsis [62][70]. The use of a zinc-polysaccharide complex has the potential to mitigate mitochondrial damage and reduce the production of cytokines induced by lipopolysaccharides (LPS) by inhibiting the MAPK signalling pathway [63][71].
Zinc is crucial for the appropriate development and function of innate and adaptive immunity and the affected signalling cascades in sepsis [64][72]. Regarding innate immunity, intracellular zinc is critical for the extravasation of neutrophils to the site of the infection and the uptake and killing of microorganisms [65][73]. Moreover, A20′s capacity to interact with ubiquitin enzyme complexes is essential for fine-tuning ubiquitin-dependent innate immune signalling pathways, including those linked to TNFR1, TLRs, IL-1R, CD40, and NOD-like receptors (NLRs). Low blood zinc, which is often found during the early stages of sepsis because of inflammation, can have negative effects on phagocytosis, oxidative burst, degranulation, cytokine production, chemotaxis [65][73], NK cell function, and lytic activity [66][74]. Zinc deficiency, both in vivo and in vitro, has been shown to result in impaired granulocyte migration and chemotaxis in response to N-formyl-L-methionyl-L-leucyl-L-phenylalanine (fMLF), as well as a reduction in the chemoattractant properties of IL-8 for T cells [65][73]. Moreover, zinc deficiency affects the modulation of the inflammatory response by IL-1ra on the cell surface [67][75]. In individuals with zinc deficiency, there is a demonstrated decrease in the exocytosis of neutrophil granules and secretory vesicles of macrophage-1 antigen (Mac-1), which is a complement receptor-3 (CR3), along with CD66b, a signal transducer involved in the adhesive activity of CD11/CD18 [65][73]. This reduction in exocytosis negatively impacts critical immune functions such as phagocytosis, oxidative burst, and granule release [68][76]. This reduction in exocytosis negatively impacts critical immune functions such as phagocytosis, oxidative burst, and granule release [69][77]. Furthermore, zinc deficiency leads to a decrease in the production of thymulin, which is essential for the differentiation and functioning of T cells [70][78]. It also impairs the production of Th1 cytokines, including TNF-α, IL-2, and IFN-γ, both in cell culture models [71][79] and in vivo [72][80].
Regarding adaptive immunity, zinc restores lymphocyte production [73][81] and natural killer (NK) activity [74][82] and promotes autophagy in vitro by maintaining the proper structure and function of zinc-dependent lysosomal enzymes, including proteases, peptidases, phosphatases, nucleases, glycosidases, sulfatases, and lipases [75][83]. In TLR- and TNFR-triggered signalling pathways, A20 was shown to downregulate the expression of IL-1β, TNF-α, CRP [50][58] and inhibit T- and B-cell-induced NF-κB signalling, interacting with proteins that bind to ubiquitin and the zinc finger domain of Inhibitor of κB kinase (IKK) gamma (IKK-γ) [12][20]. Zinc signals mediated by the zinc transporter ZIP8, a Zrt-/Irt-like protein metal transporter with multiple natural substrates, influence the adaptive immune response during bacterial pneumonia, protecting lung epithelium [76][77][84,85].
Zinc is a cofactor for many enzymes in microorganisms and can directly affect microbial growth and function, affect their virulence, and toxin production and either guarantee or disrupt key processes for survival [78][86]. It has been recently shown that pathogens attacking peripheral blood mononuclear cells disrupt their immune function, impairing phagocytosis in sepsis [79][87]. These data suggest bacterial uptake and lysosomal degradation by monocytes or macrophages during sepsis and invasive infections are inhibited, leading to excessive lysosomal load with hid to zinc bacteria or viruses into dysfunctional monocytes or macrophages [80][81][88,89]. Furthermore, zinc overexposure shifts gut microbiota to antibiotic-resistance genes [82][90] by increasing the zinc available for bacteria [83][91], interrupting the barriers to nutritional immunity [84][92]. While zinc is essential in small amounts, excessive zinc can disrupt microbial cell membranes, interfere with essential cellular processes, and ultimately lead to microbial cell death. Thus, zinc sequestration by the human immune system facilitated by interleukin IL-6 helps to intoxicate engulfed pathogens and act cytoprotectively by neutralizing ROS and nitrogen species [85][86][93,94].
Stimulation of nucleotide-binding oligomerisation domain-2 (NOD2) leads to an increase in intracellular zinc levels, which, in turn, enhances autophagy and promotes the clearance of bacteria. The regulation of metallothionein genes in human monocyte-derived macrophages (MDMs) relies on the zinc-dependent transcription factor MTF-1 [87][95]. Knocking down MTF-1 does not impact the baseline bacterial clearance by MDMs. However, when there is continuous NOD2 stimulation, the increase in intracellular zinc, autophagy, and bacterial clearance is compromised in MDMs lacking MTF-1 [87][95]. MTF-1 is involved in the transcription of genes responsible for sequestering and transporting zinc within cells and regulating intracellular signalling pathways by activating metallothioneins [88][96]. MTF-1 moves between the cytoplasm and nucleus, with its export likely mediated by its interaction with chromosomal maintenance 1 (Crm1), a major mammalian export protein facilitating the transport of large macromolecules, including RNA and proteins, across the nuclear membrane to the cytoplasm (Figure 2). The addition of zinc or induction of autophagy restores bacterial clearance in MDMs after metallothionein knockdown. In addition, in MDMs, NOD2 works synergistically with the Pattern Recognition Receptors (PRRs) TLRs 5 and 9 to enhance the effects of metallothioneins [87][95]. In mice, the intestinal microbiota plays a role in regulating the expression of metallothioneins, zinc levels, autophagy, and bacterial clearance in intestinal macrophages [87][95].
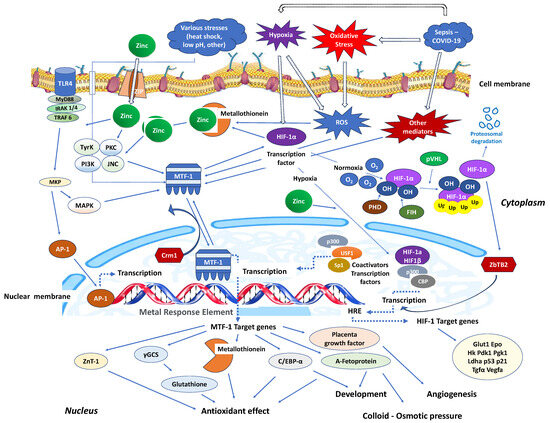
Figure 2. Critical zinc effects on transcriptional factors apart from NF-κB. MTF-1: Intracellular metal responsive transcription factor 1; HIF-1α: hypoxia-inducible factor 1-alpha; AP-1: activator protein-1; MRE: metal-response element; PHD: prolyl hydroxylase domain enzyme; FIH: factor-inhibiting HIF; Crm1: chromosomal maintenance 1; O2: oxygen molecule; HRE: hypoxia response element; p300: protein 300; CBP: CREB-binding protein; ZBTB2: zinc finger and bric-a-brac (BTB)-domain-containing protein 2; OH: hydroxyl group; Ub: ubiquitin; ZIP: Zinc importer protein; pVHL: von Hippel–Lindau protein, ROS: reactive oxygen species; MyD88: myeloid differentiation primary response 88; IRAK-4: interleukin-1 receptor-associated kinase 4; TLR: toll-like receptor; TRAF: TNF receptor-associated factor; MKP: MAP kinase phosphatases; MAKP: mitogen-activated protein kinase; USF1: upstream transcription factor 1; γGCS: γ-glutamylcysteine synthetase; Glut1: Glucose transporter 1; Hk: hexokinase; PdK1: pyruvate dehydrogenase kinase-1; PgK1: phosphoglycerate kinase-1; Ldha: lactate dehydrogenase A; Tgfa: transforming growth factor, alpha; Vegfa: vascular endothelial growth factor A; C/EBV-α: CCAAT/enhancer binding protein alpha.
Similarly, in interaction with the activating transcription factor 4 (ATF4), HIF-1 improves the immune activity of macrophages involved in sepsis [89][97]. It has been demonstrated that inhibiting glycolysis contributes to neutrophil immunosuppression during sepsis, regulated by the PI3K/Akt-HIF-1α-pathway-mediated downregulation of lactate dehydrogenase A (LDHA) [90][98]. SARS-CoV-2 ORF3a induces mitochondrial damage and the production of mitochondrial ROS, leading to increased HIF-1α expression, which subsequently facilitates SARS-CoV-2 infection and cytokine production [91][99]. The effects of HIF-1α on vasculature, metabolism, inflammation, immune response, and apoptosis contribute to lung injury in COVID-19 [92][100]. Zinc finger and bric-a-brac (BTB)-domain-containing protein 2 (ZBTB2) enhance the expression of specific HIF-1 target genes under hypoxia. HIF-1 binding to the consensus hypoxia-responsive element (HRE) sequence recruits ZBTB2 to the gene locus, increasing p300-mediated histone acetylation and enhancing gene expression under hypoxic conditions (Figure 2). In normoxia, prolyl hydroxylases (PHDs) hydroxylate HIF-1α on two proline residues in the oxygen-dependent degradation domain, triggering von Hippel–Lindau (pVHL)-mediated ubiquitination and proteasomal degradation. Concurrently, factor inhibiting HIF (FIH), an asparaginyl hydroxylase regulated similarly to PHDs in an oxygen-dependent manner, suppresses HIF-1′s transcriptional activity in normoxia by preventing co-activator recruitment. In contrast, hypoxia inhibits PHDs and stabilizes HIF-1α, allowing it to translocate into the nucleus and dimerize with constitutively expressed HIF-1β, forming an active HIF-1 complex that triggers the transcription of genes promoting glycolytic metabolism, angiogenesis, and cell survival [93][101]. Both in vitro and in vivo studies confirm that zinc promotes angiogenesis through the HIF-1α/VEGF-signalling pathway [94][102] (Figure 2).
5. Hormonal Effects
The pituitary gland boasts the most substantial zinc concentration, which augments the performance of pituitary hormones [95][103]. When there is a shortage of zinc, it leads to a deficiency in the secretion of pituitary growth hormone (GH), nerve growth factor (NGF), insulin-like growth factor 1 (IGF-1) [96][104], and hippocampal nerve growth factor 7S (NGF-7S) [97][105]. Importantly, COVID symptoms might also be due to hypothalamic–pituitary–adrenal (HPA) axis suppression induced by the virus and the immune system. The angiotensin receptor converting enzyme 2 (ACE2) expression in the zona fasciculata and zona reticularis of the adrenal cortex, injured by the identified in autopsies COVID-19 virus, could affect glucocorticoid synthesis [98][106]. ACE2 is expressed not only in the lower respiratory tract but also in the somatosensory cortex, rectal/orbital gyrus, temporal lobe, hypothalamus/thalamus, brainstem, and cerebellum. Direct viral infection of the neuronal cells in these regions occurs by the virus binding to the ACE2 receptor, resulting in demyelination and neurodegeneration and inducing a greater risk of long-term effects in some patients recovering from COVID-19 [99][107]. Zinc deficiency and genetic susceptibility also contribute to neurodegeneration [100][108]. Thus, the elevated zinc deficiency gene for the odorant metabolizing enzyme UDP-glucuronosyltransferase (UGT) has been linked to COVID-induced anosmia [101][109].
While COVID-19 can induce stress-related metabolic disturbances and zinc deficiency can impact the proper functioning of insulin receptors on cell surfaces, the exact interactions between insulin resistance and zinc in the context of COVID-19 are not fully understood. In vivo, zinc improved the endothelium-dependent vasorelaxation, reversed the reduction of guanosine 5′-triphosphate cyclohydrolase 1 and tetrahydrobiopterin, and suppressed the elevation of ROS in streptozotocin-induced diabetic mice [102][110]. Finally, epigenetic changes on the male and female reproductive organs secondary to COVID-19 exposure are not well documented, although reports indicating adverse effects of COVID-19 on male and female gametogenesis are emerging [103][111]. SARS-CoV-2 induces cytokine storm, increases ROS in the gametogenic cells, and depletes intracellular zinc, potentially resulting in oocyte and sperm damage. Furthermore, zinc deficiency mediating ROS overproduction potentiates oocyte and sperm damage, aggravating the effects of COVID-19 [103][111].