Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Sirius Huang and Version 1 by Emmanuelle C GENIN.
Mitochondrial dysfunction occurs in numerous neurodegenerative diseases, particularly amyotrophic lateral sclerosis (ALS), where it contributes to motor neuron (MN) death. Of all the factors involved in ALS, mitochondria have been considered as a major player, as secondary mitochondrial dysfunction has been found in various models and patients.
- mitochondria
- amyotrophic lateral sclerosis
- motor neuron disease
- frontotemporal dementia
- ALS genes
- CHCHD10
1. Mitochondria and ALS
Mitochondria are a double-membrane-bound organelle in most eukaryotic cells. They are composed of the outer mitochondrial membrane (OMM), the intermembrane space (IMS), the inner mitochondrial membrane (IMM) and the matrix. The main function of mitochondria is to generate ATP by oxidative phosphorylation (OXPHOS) of ADP via the electron transport chain. Mitochondria also play a major role in the maintenance of cellular homeostasis and apoptosis. Mitochondria are key regulators of calcium homeostasis, alone or in association with the ER. Physiological neuronal functions require a large amount of ATP and thus functioning mitochondria. Neuronal mitochondria are essential for neuronal function and survival.
Of all the factors involved in ALS, mitochondria have always been considered as a major player, as secondary mitochondrial dysfunction has been identified in various models and patients. Indeed, mitochondria are involved in cellular functions damaged in ALS such as calcium homeostasis, regulation of apoptosis and protein quality control [21][1]. Moreover, the identification of CHCHD10 variants in ALS patients [22][2] was the first genetic evidence that a mitochondrial defect can cause MN damage and directly links mitochondrial dysfunction to the etiology of ALS [23,24][3][4] and FTD [25][5].
Mitochondrial dysfunction occurs in numerous neurodegenerative diseases, particularly ALS, where it contributes to MN death [26][6]. Abnormal mitochondrial morphology, defects in mitochondrial dynamics, altered activities of respiratory chain enzymes and increased production of reactive oxygen species (ROS) have been described in both ALS patients and ALS models [27][7] (Figure 1).
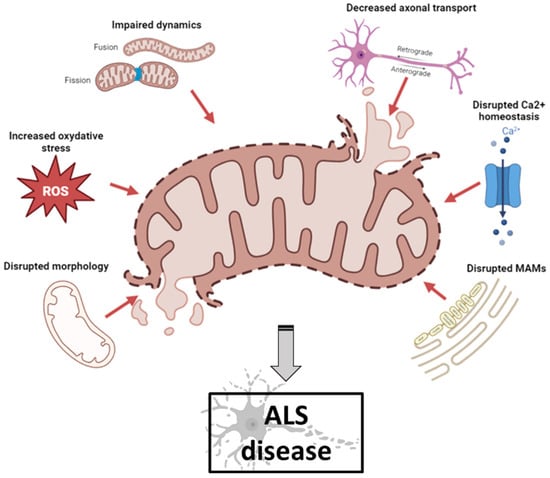
Figure 1. Schematic illustration of mitochondrial dysfunctions in ALS pathogenesis (created with biorender.com, accessed on 29 September 2023). Abnormal mitochondrial morphology, increased ROS production, defects in mitochondrial dynamics, impaired axonal transport, disruption of axonal transport and disruption of MAM integrity have been described both in ALS patients and in ALS models. ALS: amyotrophic lateral sclerosis; MAM: mitochondrial-associated membranes; ROS: reactive oxygen species.
2. Mitochondria and Ultrastructural Morphology
Mitochondrial morphology abnormalities and aggregated mitochondria were one of the first changes observed in ALS patients [28][8]. ALS MNs showed decreased mitochondrial membrane potential, impaired mitochondrial import, decreased OXPHOS and altered cristae [29,30][9][10]. Ultrastructural abnormalities of mitochondria (with vacuolated and swollen appearance, cristae abnormalities) have been associated with mitochondrial axonal transport damage. In a transgenic mouse model of fALS associated with SOD1 mutant, abnormal mitochondrial morphology and mitochondrial dysfunction in the nervous system occur early in disease progression, suggesting that these changes may predispose MNs to degeneration. Mitochondrial transport in neurons is progressively impaired early in the disease course before the onset of clinical symptoms, and retrograde transport is impaired earlier than anterograde transport [31][11].
3. Mitochondria and Reactive Oxidative Species (ROS)
In mitochondria, ROS are produced during physiological metabolism and are crucial in cellular homeostasis maintenance. Damage in OXPHOS results in a decrease in transmembrane potential and an increase in ROS production, leading directly to oxidative damage that causes mitochondrial dysfunction (generation of misfolded proteins and protein aggregates, etc.) and eventually mitophagy and apoptosis [32,33][12][13]. Oxidative stress is caused by an imbalance between oxidants and antioxidants resulting from an excess of ROS, reactive nitrogen species (RNS) or inadequate antioxidant system function. ROS contribute significantly to neuronal cell degeneration by modulating biomolecule function (DNA, RNA, lipids and proteins) and processes (nucleic acid oxidation, lipid peroxidation). Mitochondrial dysfunction and oxidative damage leading to MN degeneration have been extensively described in the pathogenesis of ALS [32][12]. Pathological mechanisms triggered directly or indirectly by ROS can lead to neuronal damage and degeneration [34][14]. MNs are extremely sensitive to oxidative stress and the central nervous system has low antioxidant capacity and low activity of protective enzymes, resulting in low cell regenerative capacity.
4. Mitochondria and Dynamics
Mitochondria form a dynamic network that is constantly dividing, fusing and changing size and shape. They constantly maintain a balance between two phenomena, fusion and fission, which are necessary for maintaining their integrity and quantity. These processes are essential for the proper functioning of these organelles. Mitochondrial fusion is the mechanism by which two mitochondria join together to form a new one. The exact event occurs by the fusion of the OMM and the IMM of the two mitochondria. Mitofusin 1 and 2 (MFN1 and MFN2) proteins are involved in fusion of the OMM, optic atrophy 1 (OPA1) protein mediates fusion of the IMM [35,36][15][16]. Mitochondria fission is the mechanism by which a mitochondrion is divided into two smaller mitochondria. This mechanism is mostly mediated by the dynamin-related protein (DRP1) and mitochondrial fission 1 (FIS1) protein. FIS1 localizes primarily on the OMM. DRP1 is a cytoplasmic protein that translocates to mitochondria and interacts with FIS1 to enhance fission. Fission takes place at ER–mitochondria junction sites and needs the oligomerization of DRP1 and several others proteins, such as FIS1 [37,38][17][18]. A positive mitochondrial membrane potential (120–200 mV) is fundamental for the physiological performance and survival of cells, especially those that have a high energy requirement. Thus, loss of mitochondrial potential membrane is an indicator of increased cell death. After fission, daughter mitochondria produced can either retain intact membrane potential or be depolarized. Depolarized mitochondria can then either return to a normal balance between fusion and fission or be eliminated by the specific autophagic process called mitophagy. Dysfunctional mitochondria show a fragmentation and perinuclear clustering, then turnover by mitophagy and degradation into lysosomes [39][19].
5. Mitochondria and Axonal Transport
Neurons are polarized cells composed of three distinct structural and functional domains: the cell body (soma), an elongated axon and the dendrites. Neurons are the longest cells and a highly structured transport machinery is needed to make them work properly. Axonal transport is essential for neuronal function, such as the maintaining of intracellular homeostasis and interaction between neighboring neurons. Transport within axons occurs in a bidirectional manner: the anterograde transport (cell body to distal axon) and the retrograde transport (distal axon to the cell body) [40][20]. Anterograde transport of newly synthesized mRNA and proteins, such as neurotransmitters, precursors and enzymes, to the distal axon terminal is required. It is mediated by kinesin movement along microtubules. Retrograde transport is necessary to maintain homeostasis by removing cytotoxic metabolites generated in the axon terminal and aged or damaged proteins and organelles targeted for degradation and recycling towards the neuron cell body [41][21]. Retrograde movement is mediated by dynein movement along microtubules.
Under physiological conditions, MNs are particularly dependent on mitochondrial ATP because of their high energy demand. Targeted transport of mitochondria from the neuronal soma to the periphery via axons is therefore essential for neuronal transmission through local ATP production. Mitochondrial dysfunction is one of the causes leading to axonal transport dysfunction. The transport and distribution of mitochondria in neurons is efficiently regulated in response to changes in neuronal activity and to a variety of physiological and pathological conditions. During axonal transport, mitochondria frequently change direction, pause or switch to permanent docking [42][22]. Specific mechanisms are necessary to transport mitochondria to their final destinations and to ensure that mitochondria remain stationary in regions of high energy demand and calcium homeostasis. In axon, mitochondria bind to motors by linking to their respective adaptor proteins, either directly or through mitochondrial receptors. Cytoplasmic dynein motors are responsible for retrograde transport of mitochondria to the soma, while KIF5 kinesin motors regulate the anterograde transport of mitochondria to distal axonal regions and synaptic terminals. Mitochondrial axonal transport is often impaired in ALS.
6. Mitochondria and Calcium Homeostasis
Mitochondria are critical for calcium homeostasis, and calcium is needed for normal transmission of signals between neurons, notably to modulate neurotransmitter release in neuromuscular junctions (NMJs). Mitochondria regulate cellular calcium ions (Ca2+) by sequestering and releasing Ca2+. Mitochondrial Ca2+ is also crucial in production of ATP, regulation of mitochondrial metabolism and cell death. Abnormalities in cellular Ca2+ signaling are common features in the pathogenesis of ALS.
Mitochondria regulate Ca2+ concentration through voltage-dependent anion-selective channel proteins (VDAC), which move Ca2+ from the matrix into the mitochondrial intermembrane space via the Ca2+ uniporter complex in the matrix [43][23]. Ca2+ release occurs at the apposition of ER and mitochondrial membranes, termed mitochondrial-associated membranes (MAMs) [44][24]. These structures control the entry of Ca2+ into the matrix of the mitochondria [45][25]. The main effectors of the Ca2+ release pathway in the ER are the inositol 1,4,5-triphosphate receptors (IP3R) and ligand-gated channels activated by the second messenger IP3 produced in response to several different extracellular signals [46][26]. The IP3R/VDAC-complex is one of the most important protein assemblies linking the ER and mitochondria [47,48][27][28]. The IP3R releases Ca2+ from the ER, and VDAC1 is responsible for Ca2+ transfer across the OMM. The interaction between the ER protein and the vesicle-associated membrane-protein-associated protein B (VAPB) and the OMM protein, protein tyrosine phosphatase interacting protein-51 (PTPIP51), facilitates IP3R-mediated Ca2+ transfer from ER to mitochondria, mitochondrial ATP production and autophagy [49,50][29][30]. Controlled Ca2+ release leads to the activation of OXPHOS activity and ATP production. When Ca2+ release is disrupted (continuous or excessive release), mitochondrial Ca2+ overload, opening of the mitochondrial permeability transition pore and triggering of the intrinsic apoptotic program are observed [51,52][31][32].
7. Mitochondria and MAM
The contact sites between the mitochondria and ER, called MAMs [53][33], are crucial for the maintenance of cellular homeostasis, including energy metabolism, Ca2+ homeostasis, cholesterol and phospholipid synthesis, ER stress response, autophagy, inflammation and mitochondrial biogenesis and transport [20,54][34][35]. MAM integrity is generally compromised in ALS [18][36] and in other neurodegenerative disorders, such as Alzheimer’s disease [55][37], Parkinson’s disease [56][38] and Huntington’s disease [57][39]. Moreover, MAM integrity is essential for mitochondrial dynamics (i.e., fission and fusion). Various proteins associated with mitochondrial dynamics are accumulated in the MAMs, such as mitofusin 2 (MFN2), interacting with apoptosis regulator protein bax to promote mitochondrial fusion [58][40]; syntaxin-17 (STX17), regulating mitochondrial fission with dynamin-related protein 1 (DRP1) [59][41]; and mitochondrial fission protein 1 (FIS1), an essential factor for mitochondrial fission, which forms a complex with B-cell-receptor-associated protein 31 (BAP31) in the MAMs [60][42]. Recently, a study reported that 16 of 21 ALS-causative genes alter the integrity of MAMs, compromising mitochondrial functions [61][43].
References
- Cozzolino, M.; Rossi, S.; Mirra, A.; Carrì, M.T. Mitochondrial Dynamism and the Pathogenesis of Amyotrophic Lateral Sclerosis. Front. Cell Neurosci. 2015, 9, 31.
- Bannwarth, S.; Ait-El-Mkadem, S.; Chaussenot, A.; Genin, E.C.; Lacas-Gervais, S.; Fragaki, K.; Berg-Alonso, L.; Kageyama, Y.; Serre, V.; Moore, D.G.; et al. A Mitochondrial Origin for Frontotemporal Dementia and Amyotrophic Lateral Sclerosis through CHCHD10 Involvement. Brain 2014, 137, 2329–2345.
- Johnson, J.O.; Glynn, S.M.; Gibbs, J.R.; Nalls, M.A.; Sabatelli, M.; Restagno, G.; Drory, V.E.; Chiò, A.; Rogaeva, E.; Traynor, B.J. Mutations in the CHCHD10 Gene Are a Common Cause of Familial Amyotrophic Lateral Sclerosis. Brain 2014, 137, e311.
- Müller, K.; Andersen, P.M.; Hübers, A.; Marroquin, N.; Volk, A.E.; Danzer, K.M.; Meitinger, T.; Ludolph, A.C.; Strom, T.M.; Weishaupt, J.H. Two Novel Mutations in Conserved Codons Indicate That CHCHD10 Is a Gene Associated with Motor Neuron Disease. Brain 2014, 137, e309.
- Sirkis, D.W.; Geier, E.G.; Bonham, L.W.; Karch, C.M.; Yokoyama, J.S. Recent Advances in the Genetics of Frontotemporal Dementia. Curr. Genet. Med. Rep. 2019, 7, 41–52.
- Lin, M.T.; Beal, M.F. Mitochondrial Dysfunction and Oxidative Stress in Neurodegenerative Diseases. Nature 2006, 443, 787–795.
- Cozzolino, M.; Carrì, M.T. Mitochondrial Dysfunction in ALS. Prog. Neurobiol. 2012, 97, 54–66.
- Sasaki, S.; Iwata, M. Mitochondrial Alterations in the Spinal Cord of Patients With Sporadic Amyotrophic Lateral Sclerosis. J. Neuropathol. Exp. Neurol. 2007, 66, 10–16.
- Genin, E.C.; Madji Hounoum, B.; Bannwarth, S.; Fragaki, K.; Lacas-Gervais, S.; Mauri-Crouzet, A.; Lespinasse, F.; Neveu, J.; Ropert, B.; Augé, G.; et al. Mitochondrial Defect in Muscle Precedes Neuromuscular Junction Degeneration and Motor Neuron Death in CHCHD10S59L/+ Mouse. Acta Neuropathol. 2019, 138, 123–145.
- Singh, T.; Jiao, Y.; Ferrando, L.M.; Yablonska, S.; Li, F.; Horoszko, E.C.; Lacomis, D.; Friedlander, R.M.; Carlisle, D.L. Neuronal Mitochondrial Dysfunction in Sporadic Amyotrophic Lateral Sclerosis Is Developmentally Regulated. Sci. Rep. 2021, 11, 18916.
- Magrané, J.; Cortez, C.; Gan, W.-B.; Manfredi, G. Abnormal Mitochondrial Transport and Morphology Are Common Pathological Denominators in SOD1 and TDP43 ALS Mouse Models. Hum. Mol. Genet. 2014, 23, 1413–1424.
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Oxidative Stress: A Key Modulator in Neurodegenerative Diseases. Molecules 2019, 24, 1583.
- Kowaltowski, A.J.; Vercesi, A.E. Mitochondrial Damage Induced by Conditions of Oxidative Stress. Free Radic. Biol. Med. 1999, 26, 463–471.
- Gandhi, S.; Abramov, A.Y. Mechanism of Oxidative Stress in Neurodegeneration. Oxidative Med. Cell. Longev. 2012, 2012, 428010.
- Merkwirth, C.; Langer, T. Mitofusin 2 Builds a Bridge between ER and Mitochondria. Cell 2008, 135, 1165–1167.
- Yapa, N.M.B.; Lisnyak, V.; Reljic, B.; Ryan, M.T. Mitochondrial Dynamics in Health and Disease. FEBS Lett. 2021, 595, 1184–1204.
- Sebastián, D.; Palacín, M.; Zorzano, A. Mitochondrial Dynamics: Coupling Mitochondrial Fitness with Healthy Aging. Trends Mol. Med. 2017, 23, 201–215.
- Friedman, J.R.; Lackner, L.L.; West, M.; DiBenedetto, J.R.; Nunnari, J.; Voeltz, G.K. ER Tubules Mark Sites of Mitochondrial Division. Science 2011, 334, 358–362.
- Chen, H.; Chan, D.C. Mitochondrial Dynamics-Fusion, Fission, Movement, and Mitophagy-in Neurodegenerative Diseases. Hum. Mol. Genet. 2009, 18, R169–R176.
- Griffin, J.W.; Price, D.L. Axonal Transport in Motor Neuron Pathology. UCLA Forum Med. Sci. 1976, 19, 33–67.
- Maday, S.; Twelvetrees, A.E.; Moughamian, A.J.; Holzbaur, E.L.F. Axonal Transport: Cargo-Specific Mechanisms of Motility and Regulation. Neuron 2014, 84, 292–309.
- Sheng, Z.-H.; Cai, Q. Mitochondrial Transport in Neurons: Impact on Synaptic Homeostasis and Neurodegeneration. Nat. Rev. Neurosci. 2012, 13, 77–93.
- Giorgi, C.; Marchi, S.; Pinton, P. The Machineries, Regulation and Cellular Functions of Mitochondrial Calcium. Nat. Rev. Mol. Cell Biol. 2018, 19, 713–730.
- Giorgi, C.; Missiroli, S.; Patergnani, S.; Duszynski, J.; Wieckowski, M.R.; Pinton, P. Mitochondria-Associated Membranes: Composition, Molecular Mechanisms, and Physiopathological Implications. Antioxid. Redox Signal. 2015, 22, 995–1019.
- Marchi, S.; Patergnani, S.; Missiroli, S.; Morciano, G.; Rimessi, A.; Wieckowski, M.R.; Giorgi, C.; Pinton, P. Mitochondrial and Endoplasmic Reticulum Calcium Homeostasis and Cell Death. Cell Calcium 2018, 69, 62–72.
- Mak, D.-O.D.; Cheung, K.-H.; Toglia, P.; Foskett, J.K.; Ullah, G. Analyzing and Quantifying the Gain-of-Function Enhancement of IP3 Receptor Gating by Familial Alzheimer’s Disease-Causing Mutants in Presenilins. PLoS Comput. Biol. 2015, 11, e1004529.
- Kania, E.; Roest, G.; Vervliet, T.; Parys, J.B.; Bultynck, G. IP3 Receptor-Mediated Calcium Signaling and Its Role in Autophagy in Cancer. Front. Oncol. 2017, 7, 140.
- Valladares, D.; Utreras-Mendoza, Y.; Campos, C.; Morales, C.; Diaz-Vegas, A.; Contreras-Ferrat, A.; Westermeier, F.; Jaimovich, E.; Marchi, S.; Pinton, P.; et al. IP3 Receptor Blockade Restores Autophagy and Mitochondrial Function in Skeletal Muscle Fibers of Dystrophic Mice. Biochim. Et Biophys. Acta (BBA)-Mol. Basis Dis. 2018, 1864, 3685–3695.
- Honrath, B.; Matschke, L.; Meyer, T.; Magerhans, L.; Perocchi, F.; Ganjam, G.K.; Zischka, H.; Krasel, C.; Gerding, A.; Bakker, B.M.; et al. SK2 Channels Regulate Mitochondrial Respiration and Mitochondrial Ca2+ Uptake. Cell Death Differ. 2017, 24, 761–773.
- Xu, H.; Guan, N.; Ren, Y.-L.; Wei, Q.-J.; Tao, Y.-H.; Yang, G.-S.; Liu, X.-Y.; Bu, D.-F.; Zhang, Y.; Zhu, S.-N. IP3R-Grp75-VDAC1-MCU Calcium Regulation Axis Antagonists Protect Podocytes from Apoptosis and Decrease Proteinuria in an Adriamycin Nephropathy Rat Model. BMC Nephrol. 2018, 19, 140.
- Morciano, G.; Giorgi, C.; Bonora, M.; Punzetti, S.; Pavasini, R.; Wieckowski, M.R.; Campo, G.; Pinton, P. Molecular Identity of the Mitochondrial Permeability Transition Pore and Its Role in Ischemia-Reperfusion Injury. J. Mol. Cell. Cardiol. 2015, 78, 142–153.
- Bonora, M.; Morganti, C.; Morciano, G.; Pedriali, G.; Lebiedzinska-Arciszewska, M.; Aquila, G.; Giorgi, C.; Rizzo, P.; Campo, G.; Ferrari, R.; et al. Mitochondrial Permeability Transition Involves Dissociation of F1FO ATP Synthase Dimers and C-ring Conformation. EMBO Rep. 2017, 18, 1077–1089.
- Rizzuto, R.; Pinton, P.; Carrington, W.; Fay, F.S.; Fogarty, K.E.; Lifshitz, L.M.; Tuft, R.A.; Pozzan, T. Close Contacts with the Endoplasmic Reticulum as Determinants of Mitochondrial Ca2+ Responses. Science 1998, 280, 1763–1766.
- Markovinovic, A.; Greig, J.; Martín-Guerrero, S.M.; Salam, S.; Paillusson, S. Endoplasmic Reticulum–Mitochondria Signaling in Neurons and Neurodegenerative Diseases. J. Cell Sci. 2022, 135, jcs248534.
- Fujimoto, M.; Hayashi, T. New Insights into the Role of Mitochondria-Associated Endoplasmic Reticulum Membrane. In International Review of Cell and Molecular Biology; Elsevier: Amsterdam, The Netherlands, 2011; Volume 292, pp. 73–117.
- Paillusson, S.; Stoica, R.; Gomez-Suaga, P.; Lau, D.H.W.; Mueller, S.; Miller, T.; Miller, C.C.J. There’s Something Wrong with My MAM; the ER–Mitochondria Axis and Neurodegenerative Diseases. Trends Neurosci. 2016, 39, 146–157.
- Area-Gomez, E.; Schon, E.A. On the Pathogenesis of Alzheimer’s Disease: The MAM Hypothesis. FASEB J. 2017, 31, 864–867.
- Gomez-Suaga, P.; Paillusson, S.; Miller, C.C.J. ER-Mitochondria Signaling Regulates Autophagy. Autophagy 2017, 13, 1250–1251.
- Maity, S.; Komal, P.; Kumar, V.; Saxena, A.; Tungekar, A.; Chandrasekar, V. Impact of ER Stress and ER-Mitochondrial Crosstalk in Huntington’s Disease. IJMS 2022, 23, 780.
- Cerqua, C.; Anesti, V.; Pyakurel, A.; Liu, D.; Naon, D.; Wiche, G.; Baffa, R.; Dimmer, K.S.; Scorrano, L. Trichoplein/Mitostatin Regulates Endoplasmic Reticulum–Mitochondria Juxtaposition. EMBO Rep. 2010, 11, 854–860.
- Arasaki, K.; Shimizu, H.; Mogari, H.; Nishida, N.; Hirota, N.; Furuno, A.; Kudo, Y.; Baba, M.; Baba, N.; Cheng, J.; et al. A Role for the Ancient SNARE Syntaxin 17 in Regulating Mitochondrial Division. Dev. Cell 2015, 32, 304–317.
- Iwasawa, R.; Mahul-Mellier, A.-L.; Datler, C.; Pazarentzos, E.; Grimm, S. Fis1 and Bap31 Bridge the Mitochondria-ER Interface to Establish a Platform for Apoptosis Induction: Fis1 Induces Apoptosis via Bap31. EMBO J. 2011, 30, 556–568.
- Sakai, S.; Watanabe, S.; Komine, O.; Sobue, A.; Yamanaka, K. Novel Reporters of Mitochondria-associated Membranes (MAM), MAMtrackers, Demonstrate MAM Disruption as a Common Pathological Feature in Amyotrophic Lateral Sclerosis. FASEB J. 2021, 35, e21688.
More