Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Alfredo Franco-Obregón and Version 2 by Jason Zhu.
Muscle function reflects muscular mitochondrial status, which, in turn, is an adaptive response to physical activity, representing improvements in energy production for de novo biosynthesis or metabolic efficiency. Differences in muscle performance are manifestations of the expression of distinct contractile-protein isoforms and of mitochondrial-energy substrate utilization. Powerful contractures require immediate energy production from carbohydrates outside the mitochondria that exhaust rapidly. Sustained muscle contractions require aerobic energy production from fatty acids by the mitochondria that is slower and produces less force.
- healthspan
- PEMF
- magnetic fields
- muscle secretome
1. Introduction
Muscle is our largest tissue mass and, as such, has evolved to modulate the regenerative capacity and metabolism of the entire body. To fulfil this role, muscle acts as a feeding tissue for the rest of the body. This feeding role of muscle is a manifestation of its secretome, a vast combination of regenerative, metabolic, anti-inflammatory and immunocompetence factors, released into the systemic circulation as either individual myokines (muscle-derived cytokines) [1][2][3][1,2,3] or vesicle-encapsulated factors [4][5][6][4,5,6]. Mechanistically, an upregulation in mitochondrial respiratory rate triggers the myokine response pathway [7][8][9][10][7,8,9,10], whereas extracellular calcium entry [11] as well as mitochondrial respiration [7] stimulate the release of muscular extracellular vesicles. Physical activity, or exercise, is the most common way to initiate the myokine [12][13][12,13] and extracellular vesicle [6] responses, which are, hence, blunted in the old and frail who are less capable of undertaking exercise [14][15][16][14,15,16]. Upon elevated muscle mitochondrial respiration induced by exercise, components of the muscle secretome travel via the bloodstream to the collateral tissues of the body that, in turn, may reciprocate with secretome responses of their own. Collateral tissue systems known to respond to the actions of the muscle secretome include the bone and joints, the immune system, the central nervous system, the digestive system, the microbiome, and, in particular, adipose tissue [3].
2. Muscle Metabolic Phenotypes
Oxidative muscles display a predilection for fatty-acid oxidation to provide energy, whereas glycolytic muscles rely largely on carbohydrates for energy production. Oxidative- and glycolytic-muscle phenotypes subserve distinct forms of contractile activity, which mirrors these distinct modes of energy production. Endurance exercise, such as distance running, predominantly recruits the participation of oxidative muscles. Oxidative muscles mediate tonic muscle contractures that require sustained mitochondrial aerobic energy production, predominantly from fatty acids. Consequently, force generation from oxidative muscle is slower and produces less power. On the other hand, resistance exercise, such as weight lifting, largely relies on the participation of glycolytic muscles. Glycolytic muscles mediate sudden bursts of contraction, requiring immediate, albeit relatively short-lived, energy production from carbohydrates, without the assistance of oxygen (anaerobically) outside of the mitochondria. Consequently, glycolytic muscles produce more powerful contractures than oxidative muscles, but exhaust more rapidly. Glycolytic muscles also accrue a substantial oxygen debt in anticipation of final mitochondrial oxidation of preliminarily catalysed carbohydrates in the form of lactic acid [17][18]. Extending from their distinct contractile signatures, oxidative and glycolytic muscles have also earned the names slow-twitch (type I) and fast-twitch (type II) muscles, respectively (Figure 1). Therefore, compared to glycolytic muscles, oxidative muscles exhibit greater mitochondrial numbers, have a higher reliance on oxidative metabolism [18][19] and are better able to support systemic insulin-sensitivity due to their predilection for fatty-acid oxidation [19][20]. Two transcriptional cascades predominantly determine the oxidative phenotype, calcium- or mitochondria-mediated.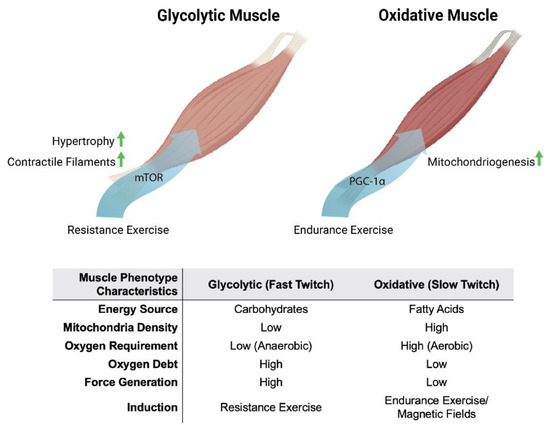
Figure 1. Metabolic and phenotypic characteristics of glycolytic and oxidative muscles in response to exercise and magnetic fields. Original figure created with BioRender.com.
3. Myoplasmic Calcium Levels
The oxidative-muscle phenotype is characterised by elevated resting calcium levels (100–300 nM), whereas glycolytic muscles are distinguished by having lower basal calcium levels (<50 nM), interspersed by high amplitude calcium transients [20][21][21,22]. These distinct calcium signatures are largely established by voltage-gated calcium entry, downstream of neuronal stimulation, and the ensuing calcium-mediated excitation–contraction coupling mechanism. In combination, these two calcium pathways serve as a developmental directive for the determination of embryonic muscle types or for the adaptive remodelling of the metabolic and functional properties of existing muscles. Mechanical loading also contributes to resting calcium levels, particularly within the functional context of oxidative muscles [20][21][21,22]. Calcium serves an enzyme catalytic role in skeletal-muscle determination. The calcium-dependent phosphatase, calcineurin (protein phosphatase 2B), is preferentially activated by the sustained calcium levels that characterise oxidative muscle [20][21][22][23][21,22,23,24]. Activated calcineurin, in turn, dephosphorylates the nuclear factor of activated T cells (NFAT), thereby allowing it to be translocated into the nucleus where it acts in cohort with other transcription factors to promote the oxidative-muscle phenotype in a calcium-sensitive manner [24][25]. Accordingly, specifically driving muscular calcineurin activity recapitulates the fast-to-slow muscle fibre switch [25][26][26,27], whereas calcineurin genetic knockdown or pharmacological inhibition of calcineurin decreases oxidative-muscle expression [21][22]. The activating calcium for calcineurin may originate from numerous sources, but often, a transient receptor potential (TRP) family member, such as TRPC1, is involved in a variety of electrically excitable cell classes [22][23][23,24].4. Mitochondrial Energy Status
The elevated respiratory activity associated with the undertaking of endurance exercise creates a deficit in mitochondrial respiratory co-factors that is manifested as increases in the ratios of AMP/ATP and NAD+/NADH. These alterations in energy status induce the expression of the transcriptional coactivator peroxisome-proliferator-activated receptor gamma coactivator-1 (PGC-1α), the master regulator of mitochondrial gene expression [27][28][28,29]. PGC-1α activity is post-translationally regulated by AMP-activated protein kinase (AMPK) phosphorylation (stimulated by a high AMP/ATP ratio) and NAD-dependent deacetylase Sirtuin–1 (Sirt1) deacetylation (stimulated by a high NAD+/NADH ratio) [27][29][30][28,30,31]. AMPK activation will also inhibit the mammalian target of rapamycin (mTOR) that will interfere with glycolytic-muscle determination [27][28][28,29]. Because of the elevated respiratory rates exhibited by oxidative muscles, muscle mitochondrial content and oxidative metabolism are enhanced, in conjunction with calcineurin signalling that consolidates the oxidative-muscle phenotype [31][32]. Therefore, it is the concomitant activation of these calcium and mitochondrial responses that establishes the genetic backdrop for the consolidation of the oxidative-muscle phenotype in response to exercise.5. Secretome-Mediated Muscle–Fat Crosstalk
Due to their greater mitochondrial and myoglobin content and denser capillary beds, oxidative fibres are also sometimes described as “red”, whereas glycolytic fibres, for comparison, are often referred to as “white” [31][32]. Generally, exercise training produces a glycolytic to oxidative switch in muscle-fibre metabolism and contractile properties [32][33][34][33,34,35]. Analogously, adipose tissue can be broadly classified as either white or brown, where white adipose is specialised for energy storage and brown adipose is adapted for energy expenditure and thermogenesis. White adipose cells are larger, contain a single large unilocular lipid droplet and tend to be proinflammatory, especially in states of prolonged physical inactivity. Brown adipose cells, on the other hand, have multiple smaller lipid droplets and greater mitochondrial content. Brown adipose cells are, hence, smaller, darker and less inflammatory than white adipose cells [35][36][36,37]. The oxidative phenotypes of both oxidative muscle and brown adipose tissue are conferred by direct and indirect exercise-induced elevations in PGC-1α, respectively [31][32], and are mutually reinforced via an interplay between the muscle and adipose secretomes [37][38]. The muscle secretome is mobilised downstream of PGC-1α activation and hence, is enhanced by exercise [7][38][7,17]. Principal amongst the tissues targeted by the muscle secretome is adipose [37][38] and key amongst the adipose-regulating factors released by exercise is irisin [38][39][40][17,39,40] (Figure 2). Irisin is responsible for the induction of spontaneous energy expenditure by adipose tissue in the form of mitochondrial uncoupled respiration, otherwise known as adaptive thermogenesis [36][41][37,41], which attenuates systemic inflammation and promotes metabolic health. Irisin descriptively “beiges” white adipose to a browner phenotype [35][36][36,37]. Muscles, hence, get redder (more oxidative) in association with a “beiging” (more thermogenic) of collateral adipose depots. Although these changes are partially reversed following periods of sedentary behaviour, epigenetic responses to exercise governing systemic metabolism and longevity have been shown to persist long after training has stopped [42] and often implicate the PGC-1α-promoter region in humans [43] and mice [44]. The exercise response is most healthful when routinely reinforced.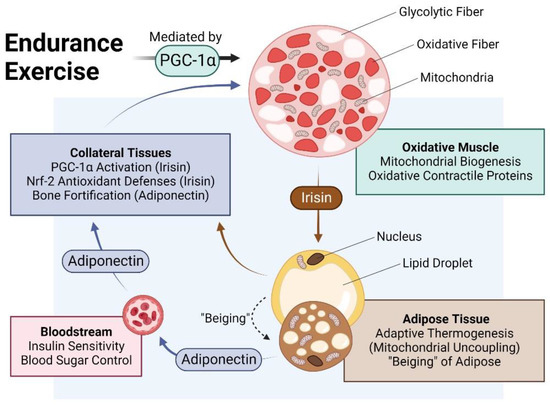
Figure 2. Impact of endurance exercise on the crosstalk between oxidative muscle and adipose tissue. Original figure created with BioRender.com.
6. Fiber Type–Fiber Size Paradox
The signalling cascades elicited for either oxidative or glycolytic determination literally compete for biosynthetic priority. As a result, muscle size and oxidative capacity are inversely related. This occurs as a consequence of the balancing of myofibrillar and mitochondrial protein production, governing contractile and metabolic adaptations, respectively [53]. Specifically, heightened mitochondrial biogenesis downstream of AMPK and PGC-1α transcriptional cascades results in an overall attenuation of protein anabolism and smaller muscle fibre size. Indeed, muscle fibres with the highest oxidative capacity have been shown to have the smallest cross-sectional area [53]. A reduction in muscle fibre cross-sectional area is hypothesised to represent a metabolic adaptation with the objective of facilitating oxygen uptake into highly oxidative fibres by effectively increasing the fibre’s surface-to-volume ratio in association with heightened mitochondrial respiratory capacity and PGC-1α activity [53][54][53,54]. Cold-water immersion is a mode of water therapy that stimulates PGC-1α activity and it exhibits key aspects of this exercise–muscle size paradox [55]. Cold-water-immersion therapy post-exercise has been generally shown to diminish resistance-training adaptations, such as muscle hypertrophy, whilst aerobic exercise performance adaptations and secretome release have been shown to be enhanced [56]. Finally, calcineurin activation is not a prerequisite for the hypertrophic response of muscle [57]. Muscular calcineurin and mitochondrial activation of PGC-1α are, hence, associated with elevated muscle metabolic capacity and reduced muscle fibre size. Although exercise is the favoured approach to enhancing oxidative-muscle development, its adoption is difficult to implement in the old, frail and infirm (also see Section 12. Is Magnetic Mitohormesis a Substitute for Exercise?). Alternative modes of inducing the oxidative-muscle phenotype in these demographics are actively being sought to help curb the growing global prevalence of metabolic syndrome and to improve the quality of life in the swelling ranks of the advanced aged.7. TRPC1 Promotes Oxidative-Muscle Development
The canonical transient receptor potential (TRPC) channel family exhibits a predilection for regulation by growth factors and is broadly involved in tissue development [58]. TRPC1 is the most ubiquitously expressed member of the family and is thought to act as a regulator of the other family members. Accordingly, TRPC1 has been implicated in diverse developmental programs, including that of oxidative muscle [18][59][19,59]. TRPC1 exhibits a capacity to heteromultimerise with the other TRPC family members, theoretically uniting their distinct activation modes into a single channel complex [60]. Mechanotransduction [60][61][62][63][60,61,62,63], magnetoreception [64][65][66][67][68][64,65,66,67,68], phototransduction [69][70][71][69,70,71], intracellular calcium sensing [72] and redox-sensing [73] are all parallel activation modes of TRPC1 that help subserve its role as an integrator of diverse forms of biophysical stimuli of consequence for development and disease [60][61][74][60,61,74], particularly with reference to skeletal muscle [75][76][77][78][79][80][81][75,76,77,78,79,80,81]. The weight of evidence indicates that TRPC1 is responsible for the elevated resting calcium levels that underlies oxidative-muscle determination. Exercise and mechanical loading promote oxidative-muscle expression, whereas physical inactivity or mechanical unloading result in oxidative-muscle loss. The elevated and sustained basal calcium levels that are required for oxidative-muscle development are diminished by physical inactivity and precede a reversal in oxidative character [20][21]. Paralleling the calcium signature of oxidative muscle, TRPC1 expression is highest in oxidative-muscle fibres [82] and wanes with disuse [18][19]. Accordingly, silencing TRPC1 expression results in oxidative-muscle loss [18][19]. TRPC1-mediated calcium entry activates the calcineurin/NFAT pathway [18][83][19,83] that, in turn, sustains TRPC1 transcription [18][84][19,84] as well as supporting PGC-1α function and oxidative-muscle maintenance [31][32]. Consequently, NFAT and TRPC1 levels decrease upon mechanical unloading and revert upon reloading, mirroring decreases and increases of oxidative-muscle expression, respectively [18][19]. Evidence thus supports cooperative roles for TRPC1, calcineurin/NFAT and PGC-1α in oxidative-muscle development, unified via mitochondrial respiration.8. Magnetic Mitohormesis
Mitochondria can be considered the stress sensors of the cell and in this capacity serve to instil survival adaptations in response to oxidative stress produced during mitochondrial responses to environmental stimuli [85]. Mitohormesis refers to an adaptive process whereby low levels of reactive oxygen species (ROS) confer the installation of survival adaptations and promote regeneration, whereas greater levels of ROS can stymie cell growth and survival [86]. Mitohormesis is a manifestation of the ability of the mitochondria to either adapt to inherent oxidative stress by enhancing their anti-oxidant defences and improved respiratory efficiency or to succumb to oxidative damage before, or beyond, adaptations. Vital in the process of integrating mitohormetic adaptations are the PGC-1α (mitochondriogenesis) and Nrf2 (anti-oxidant defences) transcriptional pathways [27][28]. It has been noted that the concomitant increases in PGC-1α and Nrf2 expression and associated decrease in insulin/IGF signalling via mTOR signalling, resulting from endurance exercise, provide the appropriate oxidative milieu to establish mitohormetic survival adaptations [27][28][28,29]. As magnetic fields stimulate mitochondrial respiration, they can be exploited as a method with which to non-invasively produce mitohormetic responses, via a novel process of magnetic mitohormesis.9. TRPC1 Confers Magnetic Mitohormesis
The enzymatic and genetic cascades activated by extremely low-frequency pulsed electromagnetic field (ELF-PEMF) exposure have recently come into clearer focus [87][88][87,88] and are commonly shown to invoke mitochondrial survival adaptations [89][90][91][89,90,91] and calcium signalling pathways [92][93][94][95][96][97][92,93,94,95,96,97] in a variety of cell classes. These two cellular response limbs need not be mutually exclusive from each other [65][90][91][98][99][100][101][65,90,91,98,99,100,101] but likely converge at the level of the mitochondria via the reciprocal and synergistic capacities of calcium to modulate mitochondrial respiration [102] and of mitochondrially-derived ROS to modulate the multimodal and integrative function of TRPC1 [73]. The manner in which the implicated calcium pathway interacts with the mitochondria most likely involves classical pathways [100], whereas the nature of magnetoreception in all likelihood entails magnetically-tuneable changes in the lifetime of a radical pair formed between a cryptochrome moiety and the mitochondrial cofactor, flavin adenine dinucleotide [87]; both mechanisms mutualistically reinforcing the other. Nonetheless, response to ELF-PEMFs has been recently shown to closely correlate with the developmental expression of TRPC1, whereas pharmacological inhibition or genetic silencing of TRPC1 precluded magnetoreception during in vitro myogenesis [65][68][102][65,68,102], chondrogenesis [64] and neurogenesis [67][103][67,103]. When specifically examined, these magnetically induced developmental responses invoke secretome activation [68][104][68,104]. Indeed, vesicular TRPC1 delivery was shown necessary and sufficient to reinstate magnetically-induced mitochondrial respiration and enhanced myogenesis in a CRISPR/Cas9 TRPC1-knockdown skeletal muscle cell line [66]. Finally, muscle cells that have been shielded from all ambient magnetic fields exhibited downregulated TRPC1 expression and reverted in vitro myogenesis [65]. Available evidence, hence, supports that TRPC1-mediated calcium entry is involved in transducing magnetic signals into diverse developmental responses by activating the calcineurin pathway (Figure 3).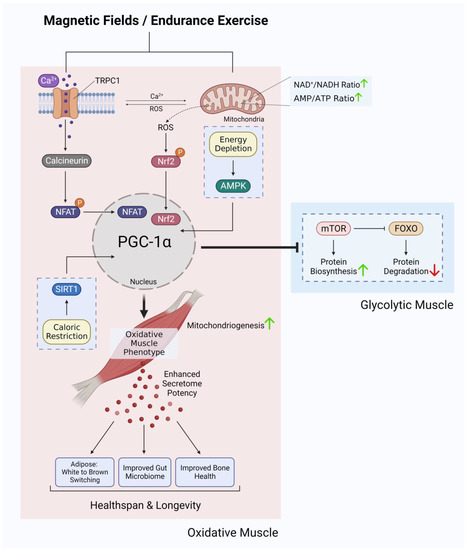
Figure 3. Ca2+ signalling and mitochondrial adaptations shared between magnetic fields and endurance training in the development of oxidative muscles. Original figure created with BioRender.com.
10. Magnetic Mitohormesis Recapitulates Oxidative-Muscle Development in Mice
10.1. Magnetic Calcineurin/NFAT Induction
In agreement with previous findings [22][23][23,24], cyclosporin and TRPC1 channel antagonists were both capable of precluding ELF-PEMF-induced in vitro myogenesis, indicating that calcineurin was being activated by TRPC1-mediated calcium entry [65]. Brief ELF-PEMF exposure also preferentially enhanced the nuclear translocation of NFATC1, whereas NFATC3 nuclear localization was reduced [65], indicating a proclivity of myogenesis towards an oxidative phenotype [105]. Accordingly, oxidative-fibre expression was increased and was associated with a decrease in oxidative-fibre cross-sectional area in mice treated weekly with ELF-PEMFs and was accompanied by enhanced running performance [106]. A decrease in the cross-sectional area of oxidative-muscle fibres has previously been reported in response to myogenin overexpression [54] and has been shown to be inversely correlated with muscle-fibre oxygen consumption rate [53]. This morphological response is explained as a physical adaptation aimed at enhancing oxygen transport in highly oxidative fibres. Provocatively, TRPC1 and mitochondria may reciprocally reinforce the response of the other via the capacity of calcium to modulate mitochondrial respiration [102] and of mitochondrially-derived ROS to modulate TRPC1 function [73]. It is intriguing to speculate that this TRPC1-mitochondrial nexus may represent a manner for the PGC-1α and calcineurin limbs of oxidative-muscle development to coordinate their developmental directives. In the exercise scenario, mechanical forces can activate TRPC1-mediated calcium entry (calcineurin induction) and central-nervous-system drive for movement will activate mitochondrial respiration (PGC-1α induction).10.2. Magnetic Modulation of Mitochondrial Energy Status
Additionally, ELF-PEMF stimulation of muscle also was shown to activate mitochondrial respiration [65]. Accordingly, the developmental adaptations observed in response to ELF-PEMF stimulation closely mirrored those commonly attributed to endurance exercise downstream of PGC-1α activation. Brief (10 min) exposure of muscle cells to low energy ELF-PEMFs was demonstrated to stimulate in vitro myogenesis in association with increased mitochondrial number, heightened mitochondrial antioxidant defences and enhanced mitochondrial-based survival adaptations [65][68][65,68]. These effects were detected in isolated muscle cells [65] as well as intact muscle [106] and were associated with transcriptional activation of PGC-1α and Nrf2, previously shown to be involved in promoting mitochondriogenesis and resistance to oxidative stress [27][107][28,107], respectively. These same transcriptional pathways are also known to be activated by endurance exercise and moderate oxidative stress [10][108][10,108]. Consistent with an increased expression of PGC-1α, mice exposed to 1 mT PEMFs for 10 min per week for several weeks [106] exhibited metabolic and functional adaptations similar to those commonly attributed to oxidative-muscle development, including enhanced resting fatty-acid oxidation (reduced respiratory exchange ratio) [109], reduced resting insulin levels [110], enhanced running performance [109] and stimulated muscle mitochondrial fatty-acid transport [111][112][111,112].11. Adipogenic Consequences of ELF-PEMF Stimulation
The adipogenic consequences of weekly ELF-PEMF treatment in mice were particularly robust and associated with elevated PGC-1α expression in both white and brown adipose samples [106]. In essence, ELF-PEMF treatment, in combination with treadmill running, increased PGC-1α most strongly in white adipose and was associated with an increase in uncoupled mitochondrial respiration (adaptive thermogenesis) and adipose mitochondrial content, as reflected by increases in the expressions of uncoupling protein 1 (Ucp1) and mitochondrial cytochrome c oxidase polypeptide 7A1 (Cox7a1), respectively [113]. These responses paralleled previously reported disparate thermogenic responses of the white versus brown adipose deposits to exercise [114]. The typically exercise-associated reddening of skeletal muscle and the browning of adipose tissue was therefore recreated in mice receiving brief weekly ELF-PEMF treatment (Figure 4).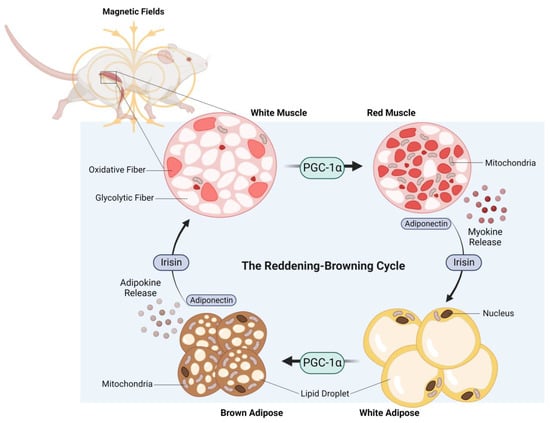
Figure 4. The reddening–browning cycle of magnetically-induced oxidative-muscle development closely mirrors the endurance-exercise-based metabolic adaptations. Original figure created with BioRender.com.
12. Is Magnetic Mitohormesis a Substitute for Exercise?
Resistance exercise, and endurance exercise to a lesser degree, produce mechanical stress and microdamage, which may serve as a stimulus to spur muscle regrowth [135][136][135,136]. In this respect, physical exercise also creates a biosynthetic regenerative energy debt that will need to be allocated towards the repair and rebuilding of damaged muscle tissue. Inadvertently, this will limit biosynthetic energy availability to implement metabolic adaptations. ELF-PEMF exposure, as it is low energy and non-mechanical, represents a more focussed form of mitochondrial stimulation. ELF-PEMF exposure may thus render stronger mitochondrial responses via the PCG-1α and Nrf2 transcriptional pathways, while producing relatively less impetus for muscle hypertrophic remodelling. It would thus be better to combine PEMF therapy with exercise, if and when possible, for greatest physiological synergism. On the other hand, mechanical stress may need to be avoided by certain frail patient demographics that, by necessity, refrain from physical exercise and consequently suffer metabolic disruption. Magnetic therapy may serve as an option for such frail demographics to maintain metabolic balance and to exploit the systemic regenerative benefits conferred by the muscle secretome. Pain and weakness are strong deterrents to exercise in the elderly [16]. Because of physical inactivity arising from such inflammatory circumstances the elder community commonly suffers from metabolic dysfunction and physical frailty. It was shown that functional mobility improved in conjunction with reduced pain in a group of elderly subjects receiving weekly ELF-PEMF treatment [133]. These results suggest that magnetic therapy may represent one manner to capacitate the elderly to undertake exercise more readily. In support of potential magnetic-therapy–exercise synergism, it was previously shown that mice, having received ELF-PEMF treatment, exhibited improved running performance after five weeks (10 min of exposure per week) [106].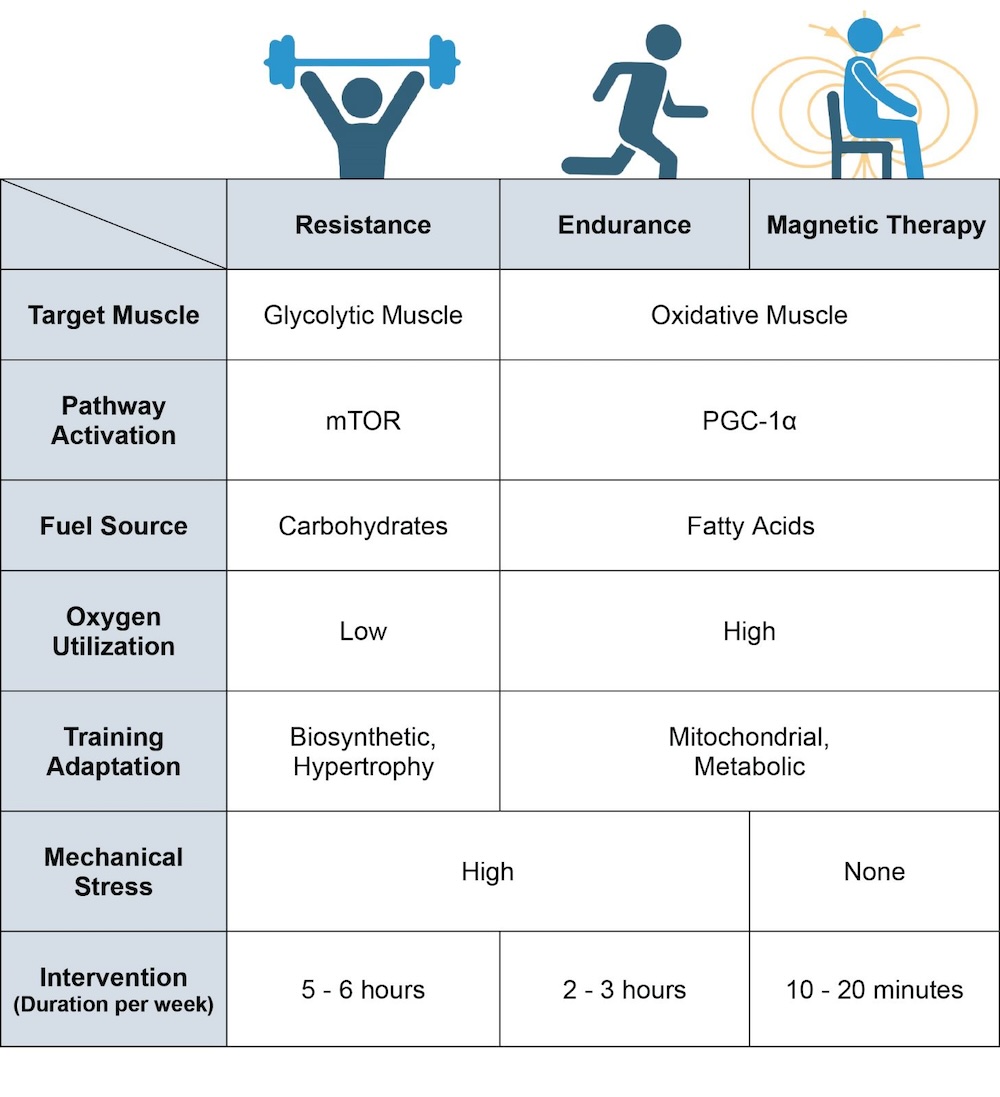 Figure 5. Breakdown of the physiological differences between resistance training, endurance training and magnetic-field intervention. Magnetic therapy refers to the use of extremely low frequency pulsed electromagnetic fields (ELF-PEMFs) such as those previously described [64][65][66][67][68][104][106][117][133][64,65,66,67,68,104,106,117,133]. Original figure created with BioRender.com.
Figure 5. Breakdown of the physiological differences between resistance training, endurance training and magnetic-field intervention. Magnetic therapy refers to the use of extremely low frequency pulsed electromagnetic fields (ELF-PEMFs) such as those previously described [64][65][66][67][68][104][106][117][133][64,65,66,67,68,104,106,117,133]. Original figure created with BioRender.com.