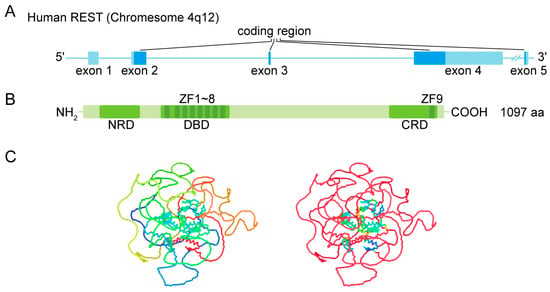
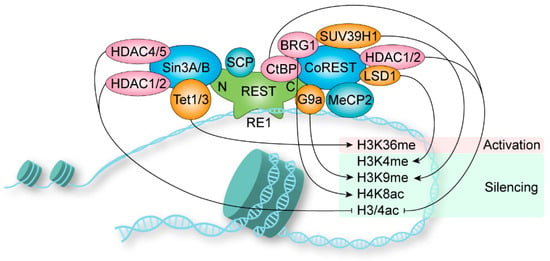
During mouse embryogenesis, REST was found to be required in repressing neuronal gene expression in both non-neural and neural precursor tissues
[14][52]. The postnatal development of the hippocampus, especially the formation of the subgranular zone (SGZ), was influenced by conditional knockout (cKO) of REST in mice
[15][53]. During early development, the loss of REST induces global hypermethylation
[16][17][54,55], and the deregulation of REST was found to be an early event in Down syndrome (DS)
[18][19][56,57]. In neural progenitor cells, REST was under the regulation of Wnt signaling, which controls various aspects of early development
[20][21][58,59].
2.2. Neuronal Differentiation
REST is a critical regulator of differentiation in embryonic stem cells (ESCs), neural stem cells (NSCs), and mature cell types
[22][23][18,51]. In ESCs, a single REST allele disruption reduced alkaline phosphatase activity and several pluripotency-associated genes expression
[24][68]. However, partial or complete loss of REST could not abrogate differentiation potential as reflected by marker gene expression
[25][69]. REST knockdown in mouse ESCs could induce neuronal lineage differentiation
[25][69], and its knockdown in human ESCs increased cell survival and the expression of mesendoderm differentiation markers
[26][70].
The repression of neuron-specific genes by REST in nonneuronal tissues highlights its crucial function during neuronal differentiation. Using ChIP-seq data mining, REST was found to mediate more than 2000 gene silencing in ESCs but not in ESC-derived neurons
[27][72]. REST is critical for maintaining the NSCs pool, and the activation of REST target genes by REST-VP16 transgene was sufficient to induce neuronal differentiation in NSCs
[28][29][73,74]. The nuclear REST was reduced during neuronal differentiation of embryonic cells, and its target genes N-methyl-D-aspartate receptor subunit 1 (NR1) and tyrosine hydroxylase (TH) were upregulated
[30][31][75,76].
2.3. Neuronal Survival
REST expression is typically low in differentiated neurons, whereas its elevation frequently exhibits protective effects. For instance, the mice with neural crest-specific KO of REST presented aberrations of the myenteric plexus, leading to neonatal lethality
[32][82]. In differentiated neural cells, overexpression of REST promoted proliferation and disrupted neurosecretion via the REST-TSC2-β-catenin signaling pathway
[33][83]. BMP/RA-inducible neural-specific protein 1 (BRINP1) was a negative regulator of cell cycle progression, and its promotor activity was inhibited by the RE1 motif within its promotor
[34][84]. REST inhibited BAF53a to quit mitosis by targeting miR-9* and miR-124 in post-mitotic neurons
[35][85].
The expression of REST in SH-SY5Y cells was induced by polychlorinated biphenyls (PCBs), and ERK2/Sp1/Sp3/REST signaling mediated the neuronal toxicity of PCBs
[36][91]. REST was increased during monosodium-glutamate-induced excitotoxicity in rats
[37][92]. REST could protect cath.-a-differentiated (CAD) neuronal cells from Mn-induced toxicity by enhancing the expression of the dopamine-synthesizing enzyme tyrosine hydroxylase (TH)
[38][93].
2.4. Neuronal Transmission and Synaptic Plasticity
REST maintains neuronal function balance by dynamically regulating the synaptic efficiency. To facilitate neurite outgrowth, REST restricted the cell adhesion molecule L1 in the embryonic nervous system
[39][94] and positively regulated the exocytosis of enlargeosomes
[40][95]. However, the constitutive expression of REST led to axon guidance mistakes in the spinal cord of chicken embryos, with the depression of the target genes N-tubulin and Ng-CAM
[41][96]. Syn1 was negatively regulated by REST to modulate synaptogenesis and neurotransmitter release
[42][97], and Sec6 has a REST binding site and function on synapse formation and synaptic plasticity
[43][98].
REST protects neurons from hyperexcitability via modulating the transcriptional activity of voltage-gated ion channel genes. REST elevated by hyperactivity inhibited Nav1.2 expression to maintain homeostasis, and silencing of REST disrupted this homeostatic response
[44][104].
2.5. Pain
REST regulates μ-opioid receptor (MOR) transcriptional activity
[45][46][47][119,120,121], contributing to MOR agonist remifentanil inducing postoperative hyperalgesia
[48][122]. PACAP was regulated by REST to mediate neuropathic pain after spinal nerve injury
[49][123]. The negative regulation of NR2B by REST was involved in bone cancer pain
[50][124]. The repression of MOR by REST reduced morphine analgesia in sarcoma-induced bone cancer pain
[51][125]. REST mediated diabetic neuropathic pain in db/db mice
[52][126]. REST also plays a major role in the acute-to-chronic pain transition upon nerve injury
[53][127]. REST KO reverted injury-induced hyperalgesia, and REST regulates peripheral somatosensory neuron programming, leading to chronic pain
[54][128].
2.6. Neuroendocrine
The cocaine- and amphetamine-regulated transcript (CART) peptide is under strict control of REST in neuroendocrine cells
[55][131]. The neurosecretory process involving dense-core vesicles (DCVs) was governed by REST in PC12 and astrocyte cells
[56][57][132,133]. The depression of REST by Kaposi’s sarcoma-associated herpesvirus (KSHV) latent open reading frame K12 (ORFK12) gene (kaposin A) regulated neuroendocrine gene expression in infected endothelial cells
[58][134].
2.7. Intelligence and Memory
Compared with mice, human-specific genes with REST occupancy were enriched in learning and memory functions
[59][60][137,138]. In early life adversity (ELA) rats, REST contributes to the memory deficits and its blockage rescued spatial memory
[61][139]. Additionally, in APP/PS1 mice, REST is essential for the maintenance of memory performance during aging
[62][140]. The elevated REST by Pb explore inhibited SV2C affected synaptic plasticity, leading to impairment of learning and memory
[63][141]. During the memory impairment induced by a flame retardant PBDE-209, REST was decreased and the inhibition of NR1 was released. CDRI-08, the ethanolic extract of a nootropic plant
Bacopa monnieri, could attenuate the impairment and restore the binding of REST to NR1 promotor
[64][142].
2.8. Aging and Alzheimer’s Disease
The REST-RE1 machinery is well maintained during aging in rats
[65][145]. By regulating autophagy and senescence in neurons, REST plays a protective role during physiological aging
[66][146]. The homologous gene of REST in
C. elegans, Spr3, is crucial for lifespan regulation, as its mutation and silencing led to shortened lifespan
[67][147]. REST was elevated during neuronal senescence induced by high glucose and palmitic acid
[68][148]. Upon peripheral nerve injury, REST was elevated in young mice but not in aged mice
[69][149].
2.9. Parkinson’s Disease
Similar to AD, REST enters the nucleus of aged dopaminergic neurons, and it is mostly absent in neurons of Parkinson’s disease (PD)
[70][153]. REST and REST4 were increased in the neurotoxin 1-methyl-4-phenyl-pyridinium ion (MPP
+) induced PD cell model
[71][154]. REST KO mice presented higher sensitivity to the dopaminergic neurotoxin MPTP
[72][155], while the neuroprotective effects of Trichostatin A (TSA) were mediated by REST in MPTP PD models
[73][156]. REST deficiency aggravated dopaminergic neurodegeneration and impaired neurogenesis in mice with MPTP-induced PD
[74][75][157,158].
2.10. Huntington’s Disease
In Huntington’s disease (HD), the increased binding of REST to RE1 reduced its target gene expression. The binding was regulated by huntingtin, and the control was lost in HD, leading to the loss of neuronal gene expression
[76][77][78][159,160,161]. The blockage of REST binding rescued BDNF
[79][162]. Huntingtin and REST formed a complex to facilitate REST translocation to the nucleus
[80][163]. The transcriptional regulation of REST by huntingtin interacting protein 1 protein interactor (HIPPI) contributed to the deregulation of transcription in HD
[81][164]. The exon-3 skipping triggered by antisense oligonucleotides (ASOs) treatment rescued neuronal genes
[82][165].
2.11. Epilepsy
The role of REST in epilepsy is controversial, and in the literature, opposite views coexist. The elevated REST during aging was abolished in patients with drug-resistant mesial temporal lobe epilepsy (MTLE)
[83][167]. REST cKO exhibited dramatically accelerated seizure progression and prolonged afterdischarge duration
[84][168]. However, several investigations suggested that REST is proepileptogenic. REST cKO mice showed higher resistance to convulsions induced by PTZ
[85][169]. The interference with the chromatin binding of REST rescued spatial memory impaired by febrile status epilepticus
[86][170]. In kainic acid-induced status epilepticus, the repression of the HCN1 channel was mediated by REST binding
[87][171], and mild inactivation of REST reduces susceptibility
[88][172]. The antiepileptic effect of 2-deoxy-d-glucose (2DG), which inhibits BDNF and its receptor TrkB to reduce the progression of kindling, was mediated by REST
[89][50], and this effect was abolished in REST cKO mice
[90][173]. The mutation of RILP could block its interaction with REST, contributing to the progressive myoclonus epilepsy progression
[91][174]. REST and REST4 controlled the expression of preprotachykinin-A (TAC1), which encodes the neuropeptide substance P
[92][93][94][175,176,177]. REST mediated the contribution of Sit1 to epileptogenesis
[95][178]. Sirt1 reduced miR-124, which targeted REST mRNA
[96][179].
2.12. Ischemia
In ischemic damage to neurons, REST appears more like a mediator. Global ischemia induced REST expression to repress GluR2 and MOR-1, and REST knockdown protected neurons from ischemia-induced death
[97][98][99][100][180,181,182,183]. Moreover, the elevated REST antagonized the CREB signaling on CART activation, leading to increased cell death
[101][184]. Pyrvinium pamoate, an activator of casein kinase 1, can suppress REST expression and rescue neurons that would otherwise undergo cell death
[102][185].
2.13. Psychiatric Disorders
REST contributes to genome-wide epigenetic changes and is implicated in schizophrenia
[103][188]. The aberrant increase of REST at the Grin2b promoter caused deficient synaptic physiology and PFC-dependent cognitive dysfunction, a hallmark of schizophrenia
[104][189]. THP2 is the rate-limiting enzyme for 5-HT, and it is under the regulation of REST
[105][190]. The mood-stabilizing drugs lithium and sodium valproate have been shown to regulate REST expression
[106][107][191,192]. miR-9 targeted REST and PDYN, and miR-9 was targeted by REST
[108][193]. REST was elevated in care-augmented rats, and its target CRH was inhibited
[109][194]. The compound C737 mimics the helical structure of REST to regulate target genes involved in neuronal function and pain modulation and reduce stress-induced weight loss in female Tupaia
[110][195]. REST signaling contributes to stress resilience
[111][196]. The improvement in diabetes mellitus-related cognitive impairment by 9-PAHSA was mediated by REST
[112][197].
3. REST in Other Systems
3.1. Heart
REST regulates heart development, structure, and function
[113][198]. During embryonic heart development, the elevated REST inhibited an embryonic cardiac gene HCN4
[114][115][116][117][199,200,201,202]. The training-induced REST contributes to the pathological heart rate adaptation to exercise training by reducing HCN4
[118][203]. REST chronically represses T channels in cardiomyocytes
[119][204]. REST also maintains low alpha 1H expression
[120][205] and regulates CACNA1H encoding alpha-subunit of Cav3.2
[121][206], thereby regulating aldosterone and cortisol production, which are independent predictors of mortality risk for heart failure patients. Gαo, an inhibitory G protein encoded by GNAO1, was regulated by REST. In several mouse models of heart failure, Gαo is decreased in the ventricles, leading to increased surface sarcolemmal L-type Ca
2+ channel activity and impairing Ca
2+ handling in ventricular myocytes, resulting in cardiac dysfunction
[122][207].
3.2. Pancreas
REST is absent in insulinoma, insulin- and glucagon-producing cells
[123][213]. Similar to neuronal differentiation, REST is expressed in pancreas progenitors and decreased in differentiated endocrine cells
[124][214]. RE1 motif was found within various genes involved in pancreas development, including pdx-1, Beta2/NeuroD, and pax4
[125][215]. Cx36 is also controlled by REST in insulin-producing cells
[126][216]. In addition, the rs2518719 SNP could alter the RE1 motif involving pancreatic neuroendocrine tumor progression
[127][217]. It was revealed that the REST silencing could induce human amniotic fluid-derived stem cells (hAFSCs) and bone marrow-derived mesenchymal stem cells (bmMSCs) to differentiate into insulin-producing cells
[128][129][218,219].
3.3. Skin
REST is expressed in skin keratinocytes, and NRP1 was found to be under its regulation to promote cell migration by blocking the inhibition of semaphorin 3A
[130][220]. During the early neural crest stage, REST is essential for melanoblast development
[131][221]. In melanoma, the decreased REST/RE1 activity leads to aberrant metabotropic glutamate receptor 1 (mGluR1) expression, whose ectopic expression in mouse melanocytes was sufficient to induce melanoma development
[132][222]. These findings provide valuable insights into the functions of REST in the skin, although much more research is needed to fully understand its role in skin biology and pathology.
3.4. Eye
The eye, being a specialized structure derived from the skin, also relies on the proper regulation of REST. In REST cKO mice generated by the Sox1-Cre allele and the floxed REST gene, REST was excised in the early neural tissues, including the lens and retinal primordia. The slightly reduced proliferation of lens epithelial cells after birth was observed, and vacuoles formed without inducing cell apoptosis. The augmented Notch signaling and the reduced lens fiber regulator genes may contribute
[133][223]. The REST-like gene in Drosophila was named Chn, and it promotes D1 for photoreceptor cell development
[134][224].
3.5. Vascular
In vascular tissue, REST was found to regulate the proliferation of vascular smooth muscle cells by targeting the K(Ca)3.1 (IKCa) potassium channel. REST was downregulated when there was cellular proliferation, indicating an inverse relationship with IKCa
[135][225]. In addition, REST declines in the medial smooth muscle of atherosclerotic carotids, and REST overexpression inhibits smooth muscle migration. Inhibition of REST and overexpression of IKCa determine smooth muscle and matrix content of atherosclerotic lesions
[136][226].
4. REST in Cancer
4.1. Nervous System Cancer
The dysregulation of REST was found in various nervous system cancers, often playing promoting roles. In neuroblastoma cells, REST was found to be reduced during differentiation
[137][227]. REST was downregulated in mouse neuroblastoma, leading to increased neurite length
[138][228]. It was found that the loss of lncRNA neuroblastoma-associated transcript-1 (NBAT-1) contributed to aggressive neuroblastoma by activating REST
[139][229]. REST is highly expressed in glioma, and its interference inhibits tumors
[140][230]. REST mediated the inhibition of proliferation of glioma by pioglitazone
[141][231]. In medulloblastoma, REST promotes cancer progression via epigenetic modification and AKT activation
[142][232]. The abnormal expression of REST in NSCs blocks their differentiation and promotes medulloblastoma formation
[143][233]. The transient expression of a competitive form of REST, REST-VP16, inhibited medulloblastoma
[144][145][234,235].
4.2. Non-Nervous System Cancer
The regulation pattern of REST varies in different types of lung cancers. In small-cell lung cancer cells (SCLCs), A splice variant of REST with a 50-bp insert predicting a truncated REST was highly expressed
[146][241]. The decreased REST permits its target gene expression, including glycine receptor alpha1 (GLRA1) subunit, migration-promoting gene ITPKA
[147][148][149][242,243,244], leading to de-regulation of AKT activation, promoting malignant progression
[150][245]. The altered REST splicing was found to mediate the anti-tumor effects of SRRM4 targeting in SCLC
[151][246]. Further, miR-9 inhibits the proliferation and migration of lung squamous cell carcinoma cells by blocking REST/EGFR signaling
[152][247]. However, in neuroendocrine lung cancer, REST mediates neuroendocrine to non-neuroendocrine fate switch to play a pro-tumorigenic role
[153][248]. REST-regulated SCG3 provides a sensitive prognostic biomarker for neuroendocrine lung cancer
[154][249].
56. Conclusion
REST is a key transcription regulator that primarily governs gene transcription via the modification of chromatin. REST plays vital roles in both physiological and pathological processes within the nervous system as well as other non-nervous systems. The imbalance of REST dynamic regulation often leads to dysplasia or diseases. Therefore, it is necessary to conduct in-depth research on its involvement in different diseases and the underlying mechanisms is imperative to gain novel insights into potential interventions for related diseases.