Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 3 by Sirius Huang and Version 2 by Sirius Huang.
Pancreatic cancer’s substantial impact on cancer-related mortality, responsible for 8% of cancer deaths and ranking fourth in the US, persists despite advancements, with a five-year relative survival rate of only 11%. The mevalonate pathway and its components play crucial roles in the development and progression of pancreatic cancer. Targeting cholesterol metabolism, particularly through the use of statins, holds promise as a therapeutic strategy.
- pancreatic cancer
- lipid metabolism
- cholesterol metabolism
- mevalonate pathway
- lipoprotein
1. Introduction
Pancreatic cancer represents currently 8% of cancer-related deaths and is the fourth leading cause in the United States [1]. The 5-year relative survival rate is still only around 11% [1]. According to the International Agency for Research on Cancer (IARC), the global number of annual new cases and pancreatic cancer-related deaths from 2020 to 2040 is predicted to increase by 70% (496,000–844,000) and 72% (466,000–801,000), respectively [2]. Deregulating cellular metabolism and immune evasion are some of the core hallmarks of cancer [3]. It is evident that cancer cells need to keep generating cellular components such as DNA, proteins, and lipids to enable rapid cell growth [4]. It has been demonstrated that the tumor-adjacent exocrine tissue exhibits upregulation of proteins related to lipid transport, which is associated with shorter post-operative survival in pancreatic cancer patients [5]. Cholesterol is an essential structural component of cell membranes and is important for physiological function [6]. 7-dehydroxycholesterol is the precursor for vitamin D and cholesterol, which itself is the key precursor for several important molecules such as bile acids as well as hormones such as glucocorticoid, mineralocorticoid, progesterone, estrogen, and testosterone [7]. Yet, growing evidence indicates that increased cholesterol flux is a common feature of cancer, and targeting the cholesterol biosynthesis pathway has been considered a promising therapeutic strategy [8].
2. The Role of the Mevalonate Pathway and De Novo Cholesterol Synthesis in Pancreatic Cancer
Acetyl coenzyme A (Acetyl-CoA) is the central molecule that participates in fatty acid synthesis as well as cholesterol biosynthesis [4]. Acetyl-CoA abundance is elevated in acinar cells of the pancreatic cancer mouse model called KC (Pdx1-Cre; lox-stop-lox-KrasG12D/+), and acetyl-CoA in the cholesterol biosynthesis pathway supports acinar-to-ductal metaplasia (ADM) formation [9]. ADM is the precursor for pancreatic intraepithelial neoplasia (PanIN), which can further progress to invasive pancreatic cancer [10]. Acetyl-CoA is also a substrate for histone acetyltransferases (HATs). Histone acetylation can lead to changes in gene expression associated with ADM formation. The oncogenic KrasG12D mutation is sufficient to promote histone H4 and histone H3 lysine 27 acetylation in acinar cells [9]. Histone acetylation marks are “written” by HATs and “read” by bromodomains cooperatively regulating transcription [11]. Bromodomain and Extra-Terminal motif (BET) protein inhibitor JQ1 prevent interaction between BET protein, acetylated histone, and transcription factors [12] and ADM formation [9]. These data emphasize the roles of acetyl-CoA and epigenetic regulations in the early stage of pancreatic carcinogenesis. Yet, the role of acetyl-CoA as a central source in cholesterol biosynthesis is also of significant importance in pancreatic cancer development, as the inhibition of cholesterol biosynthesis-associated enzymes attenuates ADM formation [9]. This chapter will summarize and discuss the critical roles of enzymes involved in the mevalonate pathway and de novo cholesterol synthesis in pancreatic cancer. In the first step, the enzyme acetoacetyl-CoA thiolase (also known as acetyl-CoA acetyltransferase, ACAT1 in mitochondria, ACAT2 in the cytosol) catalyzes a process to generate acetoacetyl-CoA (C4) from two acetyl-CoA molecules [6] (Figure 1). Sterol O-Acyltransferase 1 (encoded by the SOAT1 gene, also known as Acyl-CoA cholesterol acyltransferase), which converts excess cholesterol to inert cholesterol esters, is also known as “ACAT1”. In the current rteview articlext, however, ACAT indicates acetyl-CoA acetyltransferase but not acyl-CoA cholesterol acyltransferase. ACAT1 is activated in several cancer types. Y407 phosphorylation of the ACAT1-tetramer by epidermis growth factor (EGF) stabilizes the active ACAT1-tetramer and supports cancer cell proliferation and tumor growth [13]. ACAT1 also possesses lysine acetyltransferase activity, which acetylates pyruvate dehydrogenase (PDHA1) and pyruvate dehydrogenase phosphatase (PDP1), leading to inhibition of the pyruvate dehydrogenase complex (PDC) activity [14]. As PDC plays a key link between glycolysis and the tricarboxylic acid (TCA) cycle, inhibition of PDC activity contributes to the Warburg effect [14]. The role of ACAT2 in pancreatic cancer and cancer development has not been fully elucidated. So far, it has been shown that elevated ACAT2 gene expression is associated with radiotherapy resistance in pancreatic cancer cells [15]. Since the cytosolic ACAT2 enzyme is involved in cholesterol synthesis but the mitochondrial ACAT1 plays a more prominent role in cancer, it may be possible that cancer cells take advantage of ACAT2-mediated attenuated PDC activity rather than enhanced ACAT2 activity for cholesterol synthesis.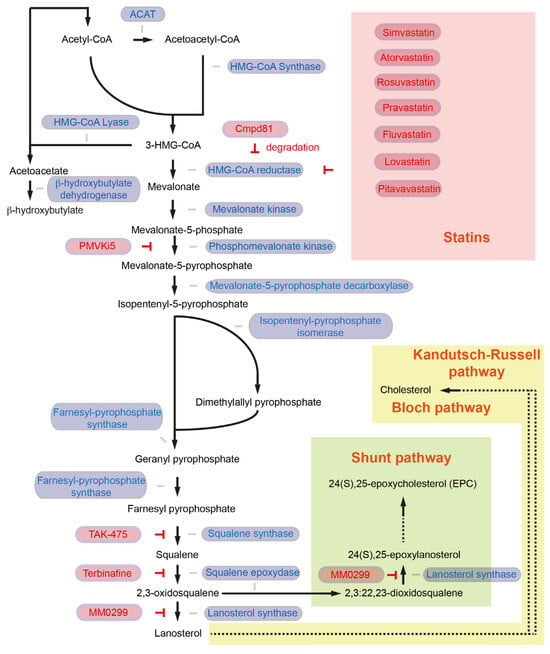
Figure 1. The cholesterol synthesis pathway. Enzymes involved in the reaction are colored in blue, and inhibitors and inhibition symbols are colored in red. Enzymes involved in ketone body biosynthesis, such as HMG-CoA lyase and β-hydroxybutylate dehydrogenase, are also included in the figure. ACAT: acetoacetyl-CoA thiolase; HMG-CoA: 3-hydroxy-3methylglutaryl-CoA.
Table 1. Inhibitors related to cholesterol metabolism.
| Intervention/Treatment | Target | Reference |
|---|---|---|
| Simvastatin | HMGCR | [25][27][55] |
| Atorvastatin | HMGCR | [26][27] |
| Rosuvastatin | HMGCR | [27] |
| Pravastatin | HMGCR | [27] |
| Fluvastatin | HMGCR | [27] |
| Lovastatin | HMGCR | [27] |
| Pitavastatin | HMGCR | [27] |
| Cmpd81 | HMGCR (degradation) | [33] |
| PMVKi5 | PMVK | [36] |
| TAK-475 | FDFT1 | [43] |
| terbinafine | SQLE | [46][48][50][51] |
| MM0299 | LSS | [54] |
| Avasimibe | SOAT1 | [56][57] |
| Surface anchor-engineered T cells with liposomal avasibime | SOAT1 | [58] |
| CP-113,818 | SOAT1 | [57] |
| K-604 | SOAT1 | [57] |
| Nilotinib | SOAT1 | [59] |
| Alirocumab | PCSK9 | [60] |
| Evolocumab | PCSK9 | [60] |
| R-IMPP | PCSK9 | [55] |
| PF-06446846 | PCSK9 | [55] |
References
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48.
- International Cancer Research Association. Available online: https://gco.iarc.fr/tomorrow/en (accessed on 1 August 2023).
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46.
- Sunami, Y.; Rebelo, A.; Kleeff, J. Lipid Metabolism and Lipid Droplets in Pancreatic Cancer and Stellate Cells. Cancers 2017, 10, 3.
- Pirhonen, J.; Szkalisity, Á.; Hagström, J.; Kim, Y.; Migh, E.; Kovács, M.; Hölttä, M.; Peränen, J.; Seppänen, H.; Haglund, C.; et al. Lipid Metabolic Reprogramming Extends beyond Histologic Tumor Demarcations in Operable Human Pancreatic Cancer. Cancer Res. 2022, 82, 3932–3949.
- Cerqueira, N.M.; Oliveira, E.F.; Gesto, D.S.; Santos-Martins, D.; Moreira, C.; Moorthy, H.N.; Ramos, M.J.; Fernandes, P.A. Cholesterol Biosynthesis: A Mechanistic Overview. Biochemistry 2016, 55, 5483–5506.
- Mayengbam, S.S.; Singh, A.; Pillai, A.D.; Bhat, M.K. Influence of cholesterol on cancer progression and therapy. Transl. Oncol. 2021, 14, 101043.
- Juarez, D.; Fruman, D.A. Targeting the Mevalonate Pathway in Cancer. Trends Cancer 2021, 7, 525–540.
- Carrer, A.; Trefely, S.; Zhao, S.; Campbell, S.L.; Norgard, R.J.; Schultz, K.C.; Sidoli, S.; Parris, J.L.D.; Affronti, H.C.; Sivanand, S.; et al. Acetyl-CoA Metabolism Supports Multistep Pancreatic Tumorigenesis. Cancer Discov. 2019, 9, 416–435.
- Storz, P. Acinar cell plasticity and development of pancreatic ductal adenocarcinoma. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 296–304.
- Marmorstein, R.; Zhou, M.M. Writers and readers of histone acetylation: Structure, mechanism, and inhibition. Cold Spring Harb. Perspect. Biol. 2014, 6, a018762.
- Shi, J.; Vakoc, C.R. The mechanisms behind the therapeutic activity of BET bromodomain inhibition. Mol. Cell 2014, 54, 728–736.
- Fan, J.; Lin, R.; Xia, S.; Chen, D.; Elf, S.E.; Liu, S.; Pan, Y.; Xu, H.; Qian, Z.; Wang, M.; et al. Tetrameric Acetyl-CoA Acetyltransferase 1 Is Important for Tumor Growth. Mol. Cell 2016, 64, 859–874.
- Goudarzi, A. The recent insights into the function of ACAT1: A possible anti-cancer therapeutic target. Life Sci. 2019, 232, 116592.
- Souchek, J.J.; Baine, M.J.; Lin, C.; Rachagani, S.; Gupta, S.; Kaur, S.; Lester, K.; Zheng, D.; Chen, S.; Smith, L.; et al. Unbiased analysis of pancreatic cancer radiation resistance reveals cholesterol biosynthesis as a novel target for radiosensitisation. Br. J. Cancer 2014, 111, 1139–1149.
- Zhou, C.; Wang, Z.; Cao, Y.; Zhao, L. Pan-cancer analysis reveals the oncogenic role of 3-hydroxy-3-methylglutaryl-CoA synthase 1. Cancer Rep. 2022, 5, e1562.
- Wang, I.H.; Huang, T.T.; Chen, J.L.; Chu, L.W.; Ping, Y.H.; Hsu, K.W.; Huang, K.H.; Fang, W.L.; Lee, H.C.; Chen, C.F.; et al. Mevalonate Pathway Enzyme HMGCS1 Contributes to Gastric Cancer Progression. Cancers 2020, 12, 1088.
- Walsh, C.A.; Akrap, N.; Garre, E.; Magnusson, Y.; Harrison, H.; Andersson, D.; Jonasson, E.; Rafnsdottir, S.; Choudhry, H.; Buffa, F.; et al. The mevalonate precursor enzyme HMGCS1 is a novel marker and key mediator of cancer stem cell enrichment in luminal and basal models of breast cancer. PLoS ONE 2020, 15, e0236187.
- Zhou, C.; Li, J.; Du, J.; Jiang, X.; Xu, X.; Liu, Y.; He, Q.; Liang, H.; Fang, P.; Zhan, H.; et al. HMGCS1 drives drug-resistance in acute myeloid leukemia through endoplasmic reticulum-UPR-mitochondria axis. Biomed. Pharmacother. 2021, 137, 111378.
- Greenspan, M.D.; Yudkovitz, J.B.; Lo, C.Y.; Chen, J.S.; Alberts, A.W.; Hunt, V.M.; Chang, M.N.; Yang, S.S.; Thompson, K.L.; Chiang, Y.C.; et al. Inhibition of hydroxymethylglutaryl-coenzyme A synthase by L-659,699. Proc. Natl. Acad. Sci. USA 1987, 84, 7488–7492.
- Zhou, C.; Wang, Z.; Yang, S.; Li, H.; Zhao, L. Hymeglusin Enhances the Pro-Apoptotic Effects of Venetoclax in Acute Myeloid Leukemia. Front. Oncol. 2022, 12, 864430.
- Gouirand, V.; Gicquel, T.; Lien, E.C.; Jaune-Pons, E.; Da Costa, Q.; Finetti, P.; Metay, E.; Duluc, C.; Mayers, J.R.; Audebert, S.; et al. Ketogenic HMG-CoA lyase and its product β-hydroxybutyrate promote pancreatic cancer progression. EMBO J. 2022, 41, e110466.
- Guillaumond, F.; Bidaut, G.; Ouaissi, M.; Servais, S.; Gouirand, V.; Olivares, O.; Lac, S.; Borge, L.; Roques, J.; Gayet, O.; et al. Cholesterol uptake disruption, in association with chemotherapy, is a promising combined metabolic therapy for pancreatic adenocarcinoma. Proc. Natl. Acad. Sci. USA 2015, 112, 2473–2478.
- Gunda, V.; Genaro-Mattos, T.C.; Kaushal, J.B.; Chirravuri-Venkata, R.; Natarajan, G.; Mallya, K.; Grandgenett, P.M.; Mirnics, K.; Batra, S.K.; Korade, Z.; et al. Ubiquitous Aberration in Cholesterol Metabolism across Pancreatic Ductal Adenocarcinoma. Metabolites 2022, 12, 47.
- Fendrich, V.; Sparn, M.; Lauth, M.; Knoop, R.; Plassmeier, L.; Bartsch, D.K.; Waldmann, J. Simvastatin delay progression of pancreatic intraepithelial neoplasia and cancer formation in a genetically engineered mouse model of pancreatic cancer. Pancreatology 2013, 13, 502–507.
- Liao, J.; Chung, Y.T.; Yang, A.L.; Zhang, M.; Li, H.; Zhang, W.; Yan, L.; Yang, G.Y. Atorvastatin inhibits pancreatic carcinogenesis and increases survival in LSL-KrasG12D-LSL-Trp53R172H-Pdx1-Cre mice. Mol. Carcinog. 2013, 52, 739–750.
- Sizar, O.; Khare, S.; Jamil, R.T.; Talati, R. Statin Medications. In StatPearls ; StatPearls Publishing: Treasure Island, FL, USA, 2023.
- Zhang, Y.; Liang, M.; Sun, C.; Qu, G.; Shi, T.; Min, M.; Wu, Y.; Sun, Y. Statin Use and Risk of Pancreatic Cancer: An Updated Meta-analysis of 26 Studies. Pancreas 2019, 48, 142–150.
- Mistafa, O.; Stenius, U. Statins inhibit Akt/PKB signaling via P2X7 receptor in pancreatic cancer cells. Biochem. Pharmacol. 2009, 78, 1115–1126.
- Mohammed, A.; Qian, L.; Janakiram, N.B.; Lightfoot, S.; Steele, V.E.; Rao, C.V. Atorvastatin delays progression of pancreatic lesions to carcinoma by regulating PI3/AKT signaling in p48Cre/+ LSL-KrasG12D/+ mice. Int. J. Cancer 2012, 131, 1951–1962.
- Uemura, N.; Hayashi, H.; Liu, Z.; Matsumura, K.; Ogata, Y.; Yasuda, N.; Sato, H.; Shiraishi, Y.; Miyata, T.; Nakagawa, S.; et al. Statins exert anti-growth effects by suppressing YAP/TAZ expressions via JNK signal activation and eliminate the immune suppression by downregulating PD-L1 expression in pancreatic cancer. Am. J. Cancer Res. 2023, 13, 2041–2054.
- Dorsch, M.; Kowalczyk, M.; Planque, M.; Heilmann, G.; Urban, S.; Dujardin, P.; Forster, J.; Ueffing, K.; Nothdurft, S.; Oeck, S.; et al. Statins affect cancer cell plasticity with distinct consequences for tumor progression and metastasis. Cell Rep. 2021, 37, 110056.
- Jiang, S.Y.; Li, H.; Tang, J.J.; Wang, J.; Luo, J.; Liu, B.; Wang, J.K.; Shi, X.J.; Cui, H.W.; Tang, J.; et al. Discovery of a Potent HMG-CoA Reductase Degrader That Eliminates Statin-Induced Reductase Accumulation and Lowers Cholesterol. Nat. Commun. 2018, 9, 5138.
- Boucher, Y.; Kamekura, M.; Doolittle, W.F. Origins and evolution of isoprenoid lipid biosynthesis in archaea. Mol. Microbiol. 2004, 52, 515–527.
- Clizbe, D.B.; Owens, M.L.; Masuda, K.R.; Shackelford, J.E.; Krisans, S.K. IDI2, a second isopentenyl diphosphate isomerase in mammals. J. Biol. Chem. 2007, 282, 6668–6676.
- Chen, Z.; Zhou, X.; Zhou, X.; Tang, Y.; Lu, M.; Zhao, J.; Tian, C.; Wu, M.; Liu, Y.; Prochownik, E.V.; et al. Phosphomevalonate Kinase Controls β-Catenin Signaling via the Metabolite 5-Diphosphomevalonate. Adv. Sci. 2023, 10, e2204909.
- Seshacharyulu, P.; Rachagani, S.; Muniyan, S.; Siddiqui, J.A.; Cruz, E.; Sharma, S.; Krishnan, R.; Killips, B.J.; Sheinin, Y.; Lele, S.M.; et al. FDPS cooperates with PTEN loss to promote prostate cancer progression through modulation of small GTPases/AKT axis. Oncogene 2019, 38, 5265–5280.
- Schmid, R.M. HMG-CoA reductase inhibitors for the treatment of pancreatic cancer. Gastroenterology 2002, 122, 565–567.
- Van de Donk, N.W.; Kamphuis, M.M.; van Kessel, B.; Lokhorst, H.M.; Bloem, A.C. Inhibition of protein geranylgeranylation induces apoptosis in myeloma plasma cells by reducing Mcl-1 protein levels. Blood 2003, 102, 3354–3362.
- Waters, A.M.; Der, C.J. KRAS: The Critical Driver and Therapeutic Target for Pancreatic Cancer. Cold Spring Harb. Perspect. Med. 2018, 8, a031435.
- Kemp, S.B.; Cheng, N.; Markosyan, N.; Sor, R.; Kim, I.K.; Hallin, J.; Shoush, J.; Quinones, L.; Brown, N.V.; Bassett, J.B.; et al. Efficacy of a Small-Molecule Inhibitor of KrasG12D in Immunocompetent Models of Pancreatic Cancer. Cancer Discov. 2023, 13, 298–311.
- Kazi, A.; Xiang, S.; Yang, H.; Chen, L.; Kennedy, P.; Ayaz, M.; Fletcher, S.; Cummings, C.; Lawrence, H.R.; Beato, F.; et al. Dual Farnesyl and Geranylgeranyl Transferase Inhibitor Thwarts Mutant KRAS-Driven Patient-Derived Pancreatic Tumors. Clin. Cancer Res. 2019, 25, 5984–5996.
- Biancur, D.E.; Kapner, K.S.; Yamamoto, K.; Banh, R.S.; Neggers, J.E.; Sohn, A.S.W.; Wu, W.; Manguso, R.T.; Brown, A.; Root, D.E.; et al. Functional Genomics Identifies Metabolic Vulnerabilities in Pancreatic Cancer. Cell Metab. 2021, 33, 199–210.e8.
- Jiang, H.; Tang, E.; Chen, Y.; Liu, H.; Zhao, Y.; Lin, M.; He, L. Squalene synthase predicts poor prognosis in stage I–III colon adenocarcinoma and synergizes squalene epoxidase to promote tumor progression. Cancer Sci. 2022, 113, 971–985.
- Bai, R.; Rebelo, A.; Kleeff, J.; Sunami, Y. Identification of prognostic lipid droplet-associated genes in pancreatic cancer patients via bioinformatics analysis. Lipids Health Dis. 2021, 20, 58.
- Wang, S.; Dong, L.; Ma, L.; Yang, S.; Zheng, Y.; Zhang, J.; Wu, C.; Zhao, Y.; Hou, Y.; Li, H.; et al. SQLE facilitates the pancreatic cancer progression via the lncRNA-TTN-AS1/miR-133b/SQLE axis. J. Cell. Mol. Med. 2022, 26, 3636–3647.
- Xu, R.; Song, J.; Ruze, R.; Chen, Y.; Yin, X.; Wang, C.; Zhao, Y. SQLE promotes pancreatic cancer growth by attenuating ER stress and activating lipid rafts-regulated Src/PI3K/Akt signaling pathway. Cell Death Dis. 2023, 14, 497.
- Zhao, F.; Huang, Y.; Zhang, Y.; Li, X.; Chen, K.; Long, Y.; Li, F.; Ma, X. SQLE inhibition suppresses the development of pancreatic ductal adenocarcinoma and enhances its sensitivity to chemotherapeutic agents in vitro. Mol. Biol. Rep. 2022, 49, 6613–6621.
- Sun, H.; Li, L.; Li, W.; Yang, F.; Zhang, Z.; Liu, Z.; Du, W. p53 transcriptionally regulates SQLE to repress cholesterol synthesis and tumor growth. EMBO Rep. 2021, 22, e52537.
- Liu, D.; Wong, C.C.; Fu, L.; Chen, H.; Zhao, L.; Li, C.; Zhou, Y.; Zhang, Y.; Xu, W.; Yang, Y.; et al. Squalene epoxidase drives NAFLD-induced hepatocellular carcinoma and is a pharmaceutical target. Sci. Transl. Med. 2018, 10, eaap9840.
- Li, C.; Wang, Y.; Liu, D.; Wong, C.C.; Coker, O.O.; Zhang, X.; Liu, C.; Zhou, Y.; Liu, Y.; Kang, W.; et al. Squalene epoxidase drives cancer cell proliferation and promotes gut dysbiosis to accelerate colorectal carcinogenesis. Gut 2022, 71, 2253–2265.
- Jun, S.Y.; Brown, A.J.; Chua, N.K.; Yoon, J.Y.; Lee, J.J.; Yang, J.O.; Jang, I.; Jeon, S.J.; Choi, T.I.; Kim, C.H.; et al. Reduction of Squalene Epoxidase by Cholesterol Accumulation Accelerates Colorectal Cancer Progression and Metastasis. Gastroenterology 2021, 160, 1194–1207.e28.
- Lasunción, M.A.; Martín-Sánchez, C.; Canfrán-Duque, A.; Busto, R. Post-lanosterol biosynthesis of cholesterol and cancer. Curr. Opin. Pharmacol. 2012, 12, 717–723.
- Nguyen, T.P.; Wang, W.; Sternisha, A.C.; Corley, C.D.; Wang, H.L.; Wang, X.; Ortiz, F.; Lim, S.K.; Abdullah, K.G.; Parada, L.F.; et al. Selective and brain-penetrant lanosterol synthase inhibitors target glioma stem-like cells by inducing 24(S),25-epoxycholesterol production. Cell Chem. Biol. 2023, 30, 214–229.e18.
- Wong, C.C.; Wu, J.L.; Ji, F.; Kang, W.; Bian, X.; Chen, H.; Chan, L.S.; Luk, S.T.Y.; Tong, S.; Xu, J.; et al. The cholesterol uptake regulator PCSK9 promotes and is a therapeutic target in APC/KRAS-mutant colorectal cancer. Nat. Commun. 2022, 13, 3971.
- Li, J.; Gu, D.; Lee, S.S.; Song, B.; Bandyopadhyay, S.; Chen, S.; Konieczny, S.F.; Ratliff, T.L.; Liu, X.; Xie, J.; et al. Abrogating cholesterol esterification suppresses growth and metastasis of pancreatic cancer. Oncogene 2016, 35, 6378–6388.
- Yang, W.; Bai, Y.; Xiong, Y.; Zhang, J.; Chen, S.; Zheng, X.; Meng, X.; Li, L.; Wang, J.; Xu, C.; et al. Potentiating the antitumour response of CD8+ T cells by modulating cholesterol metabolism. Nature 2016, 531, 651–655.
- Hao, M.; Hou, S.; Li, W.; Li, K.; Xue, L.; Hu, Q.; Zhu, L.; Chen, Y.; Sun, H.; Ju, C.; et al. Combination of metabolic intervention and T cell therapy enhances solid tumor immunotherapy. Sci. Transl. Med. 2020, 12, eaaz6667.
- Wang, Z.; Wang, M.; Zhang, M.; Xu, K.; Zhang, X.; Xie, Y.; Zhang, Y.; Chang, C.; Li, X.; Sun, A.; et al. High-affinity SOAT1 ligands remodeled cholesterol metabolism program to inhibit tumor growth. BMC Med. 2022, 20, 292.
- Liu, C.; Chen, J.; Chen, H.; Zhang, T.; He, D.; Luo, Q.; Chi, J.; Hong, Z.; Liao, Y.; Zhang, S.; et al. PCSK9 Inhibition: From Current Advances to Evolving Future. Cells 2022, 11, 2972.
More