Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Sirius Huang and Version 1 by LOU'I AL-HUSINAT.
Sepsis is a life-threatening condition that results from a profoundly dysregulated response to infection. It can lead to organ failure distant from the primary site of the infection, particularly in the kidneys. Sepsis can cause acute kidney injury (AKI). Sepsis and AKI form a vicious cycle, as sepsis is one of the main drivers of critical illness.
- norepinephrine
- sepsis
- septic shock
- septic acute kidney injury
1. Introduction
Sepsis can be defined as any dysregulated response to an infection. It can trigger septic shock, an inflammatory cascade that may involve a cytokine storm [22][1], along with circulatory system abnormalities and metabolic and cellular derangements [12][2]. Septic shock can lead to acute kidney injury (AKI), which may be life threatening or have long-term adverse sequelae [8][3]. Sepsis-associated AKI (S-AKI) is clinically distinct from AKI without sepsis. A high proportion, if not the majority, of critically ill patients develop S-AKI [23][4], which carries with it high morbidity and mortality rates [12][2]. Sepsis and AKI form a vicious cycle, as sepsis is one of the main drivers of critical illness [23][4].
The host’s response to any pathogen depends in part on the virulence of the micro-organism. The type of pathogen in sepsis helps determine the course of the infection and the outcome. The predominant organisms associated with sepsis are Staphylococcus aureus (20.5%), Pseudomonas spp. (19.9%), Enterobacteriaceae (16.0%), and fungi (19%) [24][5]. In a meta-analysis of 510 studies, gram-negative bacteremia was more closely associated with mortality than gram-positive bacteremia [25][6]. While the organism and site of infection may play a role in determining negative outcomes, approximately one-third of patients with severe sepsis do not have a positive blood culture [26][7]. Genetic factors play a role in sepsis susceptibility and poor outcomes [24][5]. Comorbidities may also play a role, as sepsis contributes to about 30% of all in-hospital cancer deaths [24][5]. The increasing incidence of sepsis and S-AKI may be due, in part, to the emergence of progressively more multidrug-resistant pathogens, longer lifespans, more urinary catheterizations, and older patients living with chronic diseases and “managed cancer” [27,28,29,30][8][9][10][11].
Sepsis leads to numerous complications, including acute respiratory distress syndrome (ARDS) [31][12] and disseminated intravascular coagulopathy (DIC), which can lead to microthrombosis, ischemic limb injury, and death [32][13]. Sepsis may also cause delirium, along with psychological or cognitive problems [33][14]. One of the most commonly reported complications of sepsis is AKI, which increases mortality risk [34][15].
2. Diagnostic Challenges in S-AKI
The Acute Dialysis Quality Initiative group published a landmark consensus definition for AKI in adults known as RIFLE, for the five stages of AKI: risk, injury, failure, loss, and end-stage disease [35][16]. AKI was characterized by a creatinine increase of ≥50% from baseline and/or a decrease in glomerular filtration rate (GFR) of ≥25% and/or decreased urinary output below a level of 0.5 mL/kg/h over at least six hours. RIFLE stated that the ≥50% increase in creatinine level occurred or could be reasonably presumed to have occurred over ≤ 7 days [35][16]. The AKI Network (AKIN) amended RIFLE by omitting the last two stages (loss and end-stage renal disease) and establishing the following stages: stage 1: risk, stage 2: injury, and stage 3: failure [36][17]. Inflammatory biomarkers, such as procalcitonin (PCT), C-reactive protein (CRP), and interleukin 18 (IL-18), are under consideration as potential diagnostic tools for S-AKI [37][18]. An increase of 0.3 mg/dL in creatinine over 48 h was part of stage 1, and the GFR criteria were removed [36][17]. Kidney Disease: Improving Global Outcomes (KDIGO) synthesized RIFLE and AKIN with some modifications [38][19].
The traditional biomarkers for AKI were urinary output, urinary indices, tubular enzymes, and cystatin C [39][20]. Biomarkers that indicate damage include neutrophil gelatinase-associated lipocalin (NGAL), kidney injury molecule-1 (KIM-1), interleukin-18 (IL-18), and liver-type fatty acid-binding proteins [37,40,41,42,43][18][21][22][23][24]. However, the evidence for some of these biomarkers is suggestive rather than conclusive, and some remain controversial. Biomarkers specific to AKI in the setting of sepsis (S-AKI) are emerging and include differentially expressed genes [44][25]. The identification of hub genes for AKI and hub genes for septic shock resulted in datasets that allowed for the identification of potential targets for future research. The genes under consideration associated with S-AKI are VMP1, SLP1, PTX3, TIMP1, OLFM4, LCN2, and S100A9 [44][25].
3. Managing S-AKI
Sepsis is managed with fluid resuscitation and early administration of antimicrobial therapy [41][22]. The CLASSIC multicenter feasibility trial explored a protocol that restricted the volume of resuscitation fluid to 151 septic shock patients and found a benefit in reducing fluids [45][26]. The CLASSIC study pointed out the potential risks of fluid overload, defined as total input minus total output divided by initial body weight. Adverse events were associated with fluid overloads over approximately 10% [46,47][27][28]. However, the results of this feasibility study remain controversial [46][27]. The 2016 Surviving Sepsis Campaign (SSC) suggests a fixed dose of crystalloid fluid (30 mL/kg) for the first three hours from diagnosis [48][29], but others recommend individualizing fluids for each patient based on their clinical status, starting with an infusion of 10 mL/kg for the first 30–60 min [49][30]. Both the 2016 SSC guidelines [50][31] and the 2018 SSC bundle [51][32] recommend early use of vasopressors, such as norepinephrine and vasopressin, in severely hypotensive septic patients, titrating the doses up until a mean arterial pressure (MAP) of at least 65 mmHg is achieved. Close clinical monitoring and regular checking of vital signs are necessary; invasive blood pressure monitoring may be considered in cases in which vasopressors are used [50][31].
4. The Pathophysiology of S-AKI
AKI is characterized by a rapidly declining glomerular filtration rate and the inability of the renal system to regulate fluid and electrolyte homeostasis [53][33]. The three main underlying causes of AKI are prerenal, postrenal, and intrinsic. Prerenal kidney injury is characterized by an abrupt reduction in blood flow to the kidney, resulting in kidney dysfunction, although the kidney itself is not damaged [54][34]. On the other hand, postrenal kidney injury occurs when the urinary tract below the kidneys is obstructed in some way, causing waste to build up in the kidneys [55][35]. S-AKI is caused by molecular patterns from pathogenic bacteria that are released from damaged cells; downstream effects are glomerular dysfunction, peritubular endothelial damage, downregulation of tubular resorption, apoptosis, and destruction of organelles from damaged cells [56][36]. Although S-AKI has been modeled as being secondary to renal ischemia, new findings from animal studies suggest that renal blood flow may paradoxically increase as renal vascular resistance decreases, so that S-AKI could occur in renal hyperemia. Ischemia need not be present for the loss of glomerular filtration [57][37]. Thus, S-AKI is a complex condition. Sepsis-induced renal microvascular dysfunction can occur, such as vasoconstriction, capillary leak syndrome, endothelial dysfunction, microthrombi, and others, leading to S-AKI [58][38]. A crucial development in S-AKI occurs when increased renal vascular resistance causes changes in the microcirculation of the renal cortex and/or renal medulla, despite normal or even elevated renal blood flow [58][38].
Historically, the pathophysiology of S-AKI was considered to be hypoperfusion and secondary tubular epithelial cell death because the most frequent causes of AKI were associated with decreased renal blood flow and ischemia [7][39]. Advances in postmortem and in vitro studies have revealed that hypoperfusion and/or ischemia are not the sole causes of S-AKI. It has been determined that hypoperfusion need not be involved in the pathogenesis of S-AKI, and S-AKI may be multifactorial in nature, with potential contributions from the inflammatory process, metabolic reprogramming, and dysregulated microcirculation [7][39].
Inflammation is the body’s primary response to invading pathogens, and a dysregulated or aberrant inflammatory response may be caused by organ dysfunction [50][31]. In sepsis, the invading pathogen releases inflammatory mediators called pathogen-associated molecular patterns (PAMPs) into the circulation. In response, damaged cells release damage-associated molecular patterns (DAMPs). Both PAMPs and DAMPs interact with pattern recognition receptors (PRRs), which are part of the body’s innate immune system [59][40]. PRR signaling leads to pro-inflammatory responses aimed at destroying or at least subduing the infectious assault [59,60][40][41]. For example, Toll-like receptors (TLRs) are a type of PRR that are expressed in renal tubular epithelial cells [61][42]. When DAMPs and PAMPs interact with TLRs in tubular epithelial cells, an imbalance in the asymmetric pro- and anti-inflammatory actions can cause a pathological increase in oxidative stress, culminating in the release of reactive oxygen species (ROS), which can damage host tissues and cause organ dysfunction [62][43]. This release of ROS can trigger inflammation in the kidney, which may be distant from the main site of the infection [7][39]. In an effort to protect itself from free radicals, which can cause necrosis, protective proteins, such as NGAL and hepcidin, are released [63,64][44][45]. Macrophages associated with H-ferritin contribute to the hepcidin-associated protective anti-inflammatory effects [65][46]. See Figure 1.
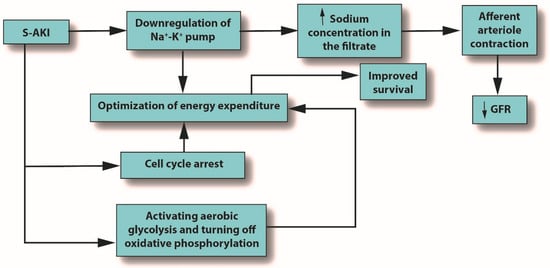
Figure 1. The outcomes of S-AKI. Note that the glomerular filtration rate is reduced only due to the influx of sodium produced by downregulation of the sodium/potassium pump; it is not an initiator. Abbreviations: GFR, glomerular filtration rate; S-AKI, septic acute kidney injury.
Severe microcirculatory dysfunction can occur in S-AKI, regardless of macrohemodynamic stability [7][39]. Alterations in the microcirculation can be caused by endothelial injury and shedding of the glycocalyx, leading to the potential formation of microthrombi due to increasing levels of white blood cells (WBCs) and platelet rolling, which eventually decrease blood flow velocity. Microcirculatory changes can also reduce GFR by several mechanisms: reduced intraglomerular hydrostatic pressure due to afferent arteriole constriction, dilatation of the efferent arteriole, intrarenal blood flow redistribution away from the medulla, and the emergence of capillaries going from afferent to efferent arterioles without passing through the glomerulus [7][39]. See Figure 2.
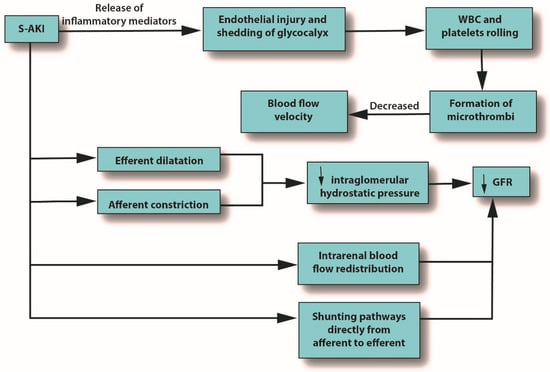
Figure 2. The role of pro-inflammatory mediators in S-AKI. Abbreviations: GFR, glomerular filtration rate; S-AKI, septic acute kidney injury; WBC, white blood cells.
Metabolic reprogramming plays a major role in the development of S-AKI and has multiple mechanisms, including cell cycle arrest, downregulation of ion transporters, and switching from aerobic glycolysis to oxidative phosphorylation. All of these mechanisms seek to optimize energy expenditure [7][39]. See Figure 3.
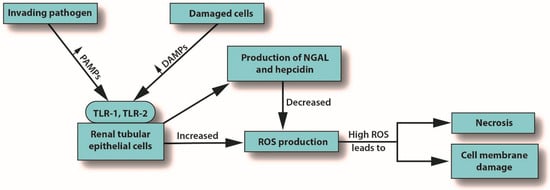
Figure 3. How S-AKI can lead to cell membrane damage and necrosis. Abbreviations: DAMPs, damaged-associated molecular patterns; NGAL, neutrophil gelatinase-associated lipocalin; PAMPs, pathogen-associated molecular patterns; ROS, reactive oxygen species; TLR, Toll-like receptor.
The inflammation caused by sepsis alone can reduce GFR, but in fact, many other mechanisms can cause or contribute to decreased GFR [66][47]. When pro-inflammatory mediators reach the glomerulus, the mesangial cells within Bowman’s capsule contract, narrowing their already narrow pores to the point that filtration is hindered [67][48]. In AKI or S-AKI, renal cells optimize their energy expenditure in response to these stresses, sometimes using the pathophysiologic system of cellular hibernation [68,69][49][50]. In cellular hibernation, the sodium/potassium ATPase pump is downregulated; normally, this pump is responsible for about 80% of renal oxygen consumption [70][51]. This causes the macula densa to detect an increase in sodium concentration in the filtrate, triggering contraction of the afferent arteriole, which, in turn, will decrease GFR [71][52].
Of course, GFR may be adversely affected by any of several mechanisms that essentially cause an imbalance between the afferent and efferent arterioles, even in the presence of increased renal blood flow to the kidneys [72][53]. This reduced GFR was found to be caused by vasodilation of the renal arteries, which lends credence to the notion that overall renal function depends on the complex interplay between systemic hemodynamics and local renal microcirculation rather than the isolated effects of each one separately [73][54].
References
- Chousterman, B.G.; Swirski, F.K.; Weber, G.F. Cytokine storm and sepsis disease pathogenesis. Semin. Immunopathol. 2017, 39, 517–528.
- Liu, J.; Xie, H.; Ye, Z.; Li, F.; Wang, L. Rates, predictors, and mortality of sepsis-associated acute kidney injury: A systematic re-view and meta-analysis. BMC Nephrol. 2020, 21, 318.
- Bagshaw, S.M.; Lapinsky, S.; Dial, S.; Arabi, Y.; Dodek, P.; Wood, G.; Ellis, P.; Guzman, J.; Marshall, J.; Parrillo, J.E.; et al. Acute kidney injury in septic shock: Clinical outcomes and impact of duration of hypo-tension prior to initiation of antimicrobial therapy. Intensive Care Med. 2009, 35, 871–881.
- Alobaidi, R.; Basu, R.K.; Goldstein, S.L.; Bagshaw, S.M. Sepsis-associated acute kidney injury. Semin. Nephrol. 2015, 35, 2–11.
- Mayr, F.B.; Yende, S.; Angus, D.C. Epidemiology of severe sepsis. Virulence 2014, 5, 4–11.
- Cohen, J.; Cristofaro, P.; Carlet, J.; Opal, S. New method of classifying infections in critically ill patients. Crit. Care Med. 2004, 32, 1510–1526.
- Rangel-Frausto, M.S. The epidemiology of bacterial sepsis. Infect. Dis. Clin. N. Am. 1999, 13, 299–312.
- Bassetti, M.; Vena, A.; Giacobbe, D.R.; Castaldo, N. Management of Infections Caused by Multidrug-resistant Gram-negative Pathogens: Recent Advances and Future Directions. Arch. Med. Res. 2021, 52, 817–827.
- Kingren, M.S.; Starr, M.E.; Saito, H. Divergent Sepsis Pathophysiology in Older Adults. Antioxid. Redox Signal. 2021, 35, 1358–1375.
- Melzer, M.; Welch, C. Does the presence of a urinary catheter predict severe sepsis in a bacteraemic cohort? J. Hosp. Infect. 2017, 95, 376–382.
- Gudiol, C.; Albasanz-Puig, A.; Cuervo, G.; Carratalà, J. Understanding and Managing Sepsis in Patients with Cancer in the Era of Antimicrobial Resistance. Front. Med. 2021, 8.
- Tongyoo, S.; Permpikul, C.; Mongkolpun, W.; Vattanavanit, V.; Udompanturak, S.; Kocak, M.; Meduri, G.U. Hydrocortisone treatment in early sepsis-associated acute respiratory distress syndrome: Results of a randomized controlled trial. Crit. Care 2016, 20, 329.
- Iba, T.; Levy, J.H. Sepsis-induced Coagulopathy and Disseminated Intravascular Coagulation. Anesthesiology 2020, 132, 1238–1245.
- Brück, E.; Schandl, A.; Bottai, M.; Sackey, P. The impact of sepsis, delirium, and psychological distress on self-rated cognitive function in ICU survivors—A prospective cohort study. J. Intensive Care 2018, 6, 1–8.
- Nandagopal, N.; Reddy, P.K.; Ranganathan, L.; Ramakrishnan, N.; Annigeri, R.; Venkataraman, R. Comparison of Epidemiology and Outcomes of Acute Kidney Injury in Critically Ill Patients with and without Sepsis. Indian J. Crit. Care Med. 2020, 24, 258–262.
- Bellomo, R.; Ronco, C.; Kellum, J.A.; Mehta, R.L.; Palevsky, P. Acute renal failure—Definition, outcome measures, animal models, fluid therapy and information technology needs: The Second International Consensus Conference of the Acute Dialysis Quality Initiative (ADQI) Group. Crit. Care 2004, 8, R204–R212.
- Mehta, R.L.; Kellum, J.A.; Shah, S.V.; Molitoris, B.A.; Ronco, C.; Warnock, D.G.; Levin, A.; Acute Kidney Injury Network. Acute Kidney Injury Network: Report of an initiative to improve outcomes in acute kidney injury. Crit. Care 2007, 11, R31.
- Xie, Y.; Huang, P.; Zhang, J.; Tian, R.; Jin, W.; Xie, H.; Du, J.; Zhou, Z.; Wang, R. Biomarkers for the diagnosis of sepsis-associated acute kidney injury: Systematic review and meta-analysis. Ann. Palliat. Med. 2021, 10, 4159–4173.
- Khwaja, A. KDIGO Clinical Practice Guidelines for Acute Kidney Injury. Nephron Clin. Pract. 2012, 120, c179–c184.
- Vijay, P.; Lal, B.B.; Sood, V.; Khanna, R.; Alam, S. Cystatin C: Best biomarker for acute kidney injury and estimation of glomerular filtration rate in childhood cirrhosis. Eur. J. Pediatr. 2021, 180, 3287–3295.
- Oh, D.-J. A long journey for acute kidney injury biomarkers. Ren. Fail. 2020, 42, 154–165.
- Gavelli, F.; Castello, L.M.; Avanzi, G.C. Management of sepsis and septic shock in the emergency department. Intern. Emerg. Med. 2021, 16, 1649–1661.
- Bonventre, J.V. Kidney Injury Molecule-1 (KIM-1): A specific and sensitive biomarker of kidney injury. Scand. J. Clin. Lab. Investig. 2008, 68, 78–83.
- Zhang, J.; Changan, W.; Liu, J.; Liang, B.; Wang, X.; Wang, C. Clinical significance of novel biomarker NGAL in early diagnosis of acute renal injury. Exp. Ther. Med. 2017, 14, 5017–5021.
- Tang, Y.; Yang, X.; Shu, H.; Yu, Y.; Pan, S.; Xu, J.; Shang, Y. Bioinformatic analysis identifies potential biomarkers and therapeutic targets of septic-shock-associated acute kidney injury. Hereditas 2021, 158, 13.
- Hjortrup, P.B.; The CLASSIC Trial Group; Haase, N.; Bundgaard, H.; Thomsen, S.L.; Winding, R.; Pettilä, V.; Aaen, A.; Lodahl, D.; Berthelsen, R.E.; et al. Restricting volumes of resuscitation fluid in adults with septic shock after initial management: The CLASSIC randomised, parallel-group, multicentre feasibility trial. Intensive Care Med. 2016, 42, 1695–1705.
- Watson, X.; Cecconi, M. Liberal or restrictive dilemma—That’s a CLASSIC! Ann. Transl. Med. 2017, 5 (Suppl. S1), S7.
- Bouchard, J.; Soroko, S.B.; Chertow, G.M.; Himmelfarb, J.; Ikizler, T.A.; Paganini, E.P.; Mehta, R.L. Fluid accumulation, survival and recovery of kidney function in critically ill pa-tients with acute kidney injury. Kidney Int. 2009, 76, 422–427.
- Evans, L.; Rhodes, A.; Alhazzani, W.; Antonelli, M.; Coopersmith, C.M.; French, C.; Machado, F.R.; Mcintyre, L.; Ostermann, M.; Prescott, H.C.; et al. Surviving sepsis campaign: International guidelines for management of sepsis and septic shock 2021. Intensive Care Med. 2021, 47, 1181–1247.
- Jozwiakì, M.; Hamzaoui, O.; Monnet, X.; Teboul, J.-L. Fluid resuscitation during early sepsis: A need for individualization. Minerva Anestesiol. 2018, 84, 987–992.
- Rhodes, A.; Evans, L.E.; Alhazzani, W.; Levy, M.M.; Antonelli, M.; Ferrer, R.; Kumar, A.; Sevransky, J.E.; Sprung, C.L.; Nunnally, M.E.; et al. Surviving Sepsis Campaign: International Guidelines for Management of Sepsis and Septic Shock: 2016. Intensive Care Med. 2017, 43, 304–377.
- Levy, M.M.; Evans, L.E.; Rhodes, A. The Surviving Sepsis Campaign Bundle: 2018 update. Intensive Care Med. 2018, 44, 925–928.
- Shah, S.R.; Tunio, S.A.; Arshad, M.H.; Moazzam, Z.; Noorani, K.; Feroze, A.M.; Shafquat, M.; Hussain, H.S.; Jeoffrey, S.A. Acute Kidney Injury Recognition and Management: A Review of the Literature and Current Evidence. Glob. J. Health Sci. 2015, 8, 120–124.
- Macedo, E.; Mehta, R.L. Prerenal failure: From old concepts to new paradigms. Curr. Opin. Crit. Care 2009, 15, 467–473.
- Makris, K.; Spanou, L. Acute Kidney Injury: Definition, Pathophysiology and Clinical Phenotypes. Clin. Biochem. Rev. 2016, 37, 85–98.
- Mårtensson, J.; Bellomo, R. Pathophysiology of Septic Acute Kidney Injury. Contrib. Nephrol. 2016, 187, 36–46.
- Bellomo, R.; Wan, L.; Langenberg, C.; May, C. Septic Acute Kidney Injury: New Concepts. Nephron Exp. Nephrol. 2008, 109, e95–e100.
- Bouglé, A.; Duranteau, J. Pathophysiology of Sepsis-Induced Acute Kidney Injury: The Role of Global Renal Blood Flow and Renal Vascular Resistance. Contrib. Nephrol. 2011, 174, 89–97.
- Peerapornratana, S.; Manrique-Caballero, C.L.; Gómez, H.; Kellum, J.A. Acute kidney injury from sepsis: Current concepts, epidemiology, pathophysiology, prevention and treatment. Kidney Int. 2019, 96, 1083–1099.
- Amarante-Mendes, G.P.; Adjemian, S.; Branco, L.M.; Zanetti, L.C.; Weinlich, R.; Bortoluci, K.R. Pattern Recognition Receptors and the Host Cell Death Molecular Machinery. Front. Immunol. 2018, 9, 2379.
- McKernan, D.P. Pattern recognition receptors as potential drug targets in inflammatory disorders. Adv. Protein Chem. Struct. Biol. 2020, 119, 65–109.
- Chen, F.; Zou, L.; Williams, B.; Chao, W. Targeting Toll-Like Receptors in Sepsis: From Bench to Clinical Trials. Antioxid. Redox Signal 2021, 35, 1324–1339.
- Kumar, V. Toll-like receptors in sepsis-associated cytokine storm and their endogenous negative regulators as future immunomodulatory targets. Int. Immunopharmacol. 2020, 89 Pt B, 107087.
- Zeng, C.; Chen, Q.; Zhang, K.; Chen, Q.; Song, S.; Fang, X. Hepatic hepcidin protects against polymicrobial sepsis in mice by regu-lating host iron status. Anesthesiology 2015, 122, 374–386.
- Qiu, Z.-L.; Yan, B.-Q.; Zhao, R.; Xu, D.-W.; Shen, K.; Deng, X.-Q.; Lu, S.-Q. Combination of hepcidin with neutrophil gelatinase-associated lipocalin for prediction of the development of sepsis-induced acute kidney injury. Clin. Chim. Acta 2021, 523, 38–44.
- Scindia, Y.; Wlazlo, E.; Leeds, J.; Loi, V.; Ledesma, J.; Cechova, S.; Ghias, E.; Swaminathan, S. Protective Role of Hepcidin in Polymicrobial Sepsis and Acute Kidney Injury. Front. Pharmacol. 2019, 10, 615.
- Frische, S. Glomerular filtration rate in early diabetes: Ongoing discussions of causes and mechanisms. J. Nephrol. 2011, 24, 537–540.
- Stockand, J.D.; Sansom, S.C. Regulation of filtration rate by glomerular mesangial cells in health and diabetic renal disease. Am. J. Kidney Dis. 1997, 29, 971–981.
- Jani, A.; Martin, S.L.; Jain, S.; Keys, D.; Edelstein, C.L. Renal adaptation during hibernation. Am. J. Physiol. Physiol. 2013, 305, F1521–F1532.
- Ratigan, E.D.; McKay, D.B. Exploring principles of hibernation for organ preservation. Transplant. Rev. 2016, 30, 13–19.
- Castillo, J.P.; Rui, H.; Basilio, D.; Das, A.; Roux, B.; Latorre, R.; Bezanilla, F.; Holmgren, M. Mechanism of potassium ion uptake by the Na+/K+-ATPase. Nat. Commun. 2015, 6, 7622.
- Bell, P.D.; Lapointe, J.Y.; Peti-Peterdi, J. Macula Densa Cell Signaling. Annu. Rev. Physiol. 2003, 65, 481–500.
- Inscho, E.W.; Imig, J.D.; Cook, A.K. Afferent and Efferent Arteriolar Vasoconstriction to Angiotensin II and Norepinephrine In-volves Release of Ca2+ from Intracellular Stores. Hypertension 1997, 29, 222–227.
- Jamshidi, P.; Najafi, F.; Mostafaei, S.; Shakiba, E.; Pasdar, Y.; Hamzeh, B.; Moradinazar, M. Investigating associated factors with glomerular filtration rate: Structural equation modeling. BMC Nephrol. 2020, 21, 30.
More