Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 3 by Jessie Wu and Version 2 by Prashant Kaushik.
Alzheimer’s disease, a devastating neurodegenerative disorder, poses a significant challenge for early diagnosis and treatment. In the quest to uncover biomarkers that could aid in early detection, proteomics techniques have emerged as powerful tools with immense potential.
- Alzheimer’s disease
- biomarkers
- proteomics
- microarray
- bioinformatics
1. Gel-Based Quantitative Technique and Differential Proteomics
Gel electrophoresis serves as a laboratory method employed to segregate and examine macromolecules, relying on their size and charge differentials by implementing an electric field through a gel matrix [37][1]. Widely applied in molecular biology and genetics, this technique facilitates the analysis of DNA, RNA, and proteins. In the realm of proteomics, it plays a pivotal role in identifying biomarkers, mainly concentrating on two variants: 2D PAGE and SDS-PAGE. The 2D-PAGE method combines two separation dimensions for higher resolution. It involves separating proteins from biological samples based on their isoelectric point and size using polyacrylamide gel electrophoresis [38][2]. Within the realm of neuroproteomics, conventional investigations have often utilized 2-DE and 2D-DIGE as the primary methods for comparing protein patterns across varying scenarios [39][3]. Over the years, multiple methods have emerged to expand and improve 2D-PAGE, resulting in a reliable and consistent approach. The implementation of immobilized pH gradient (IPG) strips, a replacement for tube gels with ampholytes, has successfully eradicated the problem of ‘cathodic drift’ during isoelectric focusing (IEF). This advancement has led to a notable increase in the reproducibility of samples, making the technique more reliable for proteomic studies. The development of narrow pH ranges in IPG strips (e.g., 4–7, 5–8) enables better separation of proteins with similar isoelectric points than traditional broad pH range strips (e.g., 3–10) [40][4]. However, solubilization remains a challenge in proteomics since ionic detergents like SDS (used in SDS-PAGE) cannot be used for lipidated proteins, like transmembrane proteins, due to interference with the focusing process in IEF [41][5]. SDS has been used in some cases, but dialysis of samples before IEF is necessary. However, this poses a limitation when working with precious biological samples due to potential sample loss. In response to this challenge, chaotropic agents like urea and thiourea, in conjunction with zwitterion detergents (e.g., CHAPS) have been utilized to avoid protein precipitation during both IEF and SDS-PAGE [42][6]. Tributyl phosphine has been used as a reducing agent instead of dithiothreitol [43][7]. Despite advancements in 2D-PAGE techniques, challenges and limitations remain. Solubilizing methods are predominantly constrained to cytosolic proteins, posing difficulties in generating gel maps for membranous and lipidated proteins. Quantification of protein alterations relies on 2D image analysis software, often demanding replicates for comparison and spot alignment. To address inter-gel disparities, 2D-DIGE employs fluorescent cyanine dyes (Cy2–Cy5) for distinct sample labeling, enabling their consolidation and simultaneous electrophoresis in a single 2D gel [44][8]. Within the context of 2D-DIGE, it becomes possible to quantify individual spots on a single gel, and the alignment of multiple gels can be achieved through referencing an internal standard labeled with Cy2 [45][9]. According to Naseri’s report in 2020, 2D-DIGE identified two client proteins, SNAP-25, and dynamin-1, suggesting that abnormal protein palmitoylation may play a crucial role in the development of ND [46][10].
2. Mass Spectrometry (MS) for Protein Identification and Quantification
In recent years, there has been a notable shift from 2D gel-based approaches to MS-based neuroproteomics studies [47][11]. The shift is primarily steered by progress in MS instrument design, the integration of resilient quantitative MS methods, adaptation for working with limited protein quantities in neuroproteomics research, and the increasing fascination with exploring complex membrane proteins that pose challenges for analysis through traditional 2D gel techniques [48][12]. Even as MS-based investigations employing stable isotope labels or label-free methodologies gain traction, gel-based techniques remain prevalent owing to their accessibility and straightforward preparation, particularly when dealing with a restricted number of samples. After detecting protein spots of interest, the proteins must be eluted from the gel for MS analysis. Usually, the spots corresponding to the desired protein(s) are apart from the gel and undergo diverse treatments and chemical alterations to aid protein fragmentation by a protease, resulting in the formation of multiple peptides [49][13]. The process of peptide mass fingerprinting involves obtaining characteristic mass fingerprints of a protein by analyzing the smaller peptides resulting from protease cleavage, and their molecular weights are determined using MS Figure 1 [50][14]. In this method, MS analysis is used to determine the experimental masses. Subsequently, a database search is conducted where the experimental masses are compared to in silico ‘digestion’ generating protein-specific mass fingerprints. The identification of the protein of interest is determined based on the quality of peptide matches. Mass spectrometry is indispensable for protein identification and proteomic analysis. Before the development of ‘softer’ ionization techniques, protein identification relied on specific antibodies or Edman degradation and protein sequencing, which required educated guesses based on molecular weight and pI knowledge [51][15]. Edman degradation and database searching are lengthy and labor-intensive methods. The two most common techniques for MS analysis of proteins are matrix-assisted laser desorption ionization (MALDI) and electrospray ionization (ESI) [52][16]. MALDI involves ionizing proteins using a laser in a time-of-flight mass spectrometer, providing precise mass-to-charge ratio determination. It offers high-throughput capabilities, simplifies sample preparation, and allows direct analysis of intact proteins and peptides from complex samples. Comparing mass spectra from control and disease samples helps identify differentially expressed proteins, making MALDI-TOF a pivotal tool for biomarker discovery and disease understanding. MALDI-TOF MS is highly valuable for peptide mass fingerprinting (PMF), a well-established protein identification method [53][17]. PMF entails the comparison of a distinctive list of peptide masses generated by targeted protein cleavage with a computer-simulated list of peptides resulting from the digestion of established protein sequences stored in databases [54][18]. To confirm protein identification, tandem spectrometry (MS/MS) can be used with various search engines (e.g., Mascot, ProFound, Sonar, MS-Fit, Sequest) that offer simple, fast, and automated protein identification (MS and/or MS/MS search). Nonetheless, it is crucial to take into account that the acquired outcomes are statistical in nature and to be mindful of the constraints inherent in the statistical identification approach. As noted by [55][19], blood emerges as an appealing avenue for biomarker exploration due to its ease of access and the existence of proteins originating not solely from blood but also from other tissues, courtesy of its systemic circulation within the body [56][20]. Blood-found proteins provide valuable insights into the organism’s health status. With MALDI-TOF MS, there is no need for pre-selecting biomarker candidates as multiple compounds can be analyzed in one experiment, offering a significant advantage [57][21]. For slowly progressing disorders like AD, identifying and monitoring biomarkers is crucial for evaluating the efficacy of disease-modifying drugs. Biomarker study data from clinical trials inform decisions on drug progression. Researchers investigate strategies such as γ-secretase inhibitors against βA and cholinergic system therapies. Portelius et al. used MALDI-TOF MS with immunoprecipitation, revealing that shorter βA isoforms (e.g., βA (1–14), βA (1–15)) are more γ-secretase inhibitor-responsive than longer ones (e.g., βA (1–40) or βA (1–42)) [58][22]. The combination of MS and immunoprecipitation for βA analysis proves essential in targeted βA proteomics. MS enables accurate identification and verification of proteins and antigens. In 1993, the first targeted βA proteomics using MALDI-TOF MS detected multiple βA isoforms in CSF. New findings from MALDI-TOF MS studies have exposed an alternative pathway for APP (Amyloid Precursor Protein) cleavage, with α- and β-secretase playing a role distinct from γ-secretase [59][23].
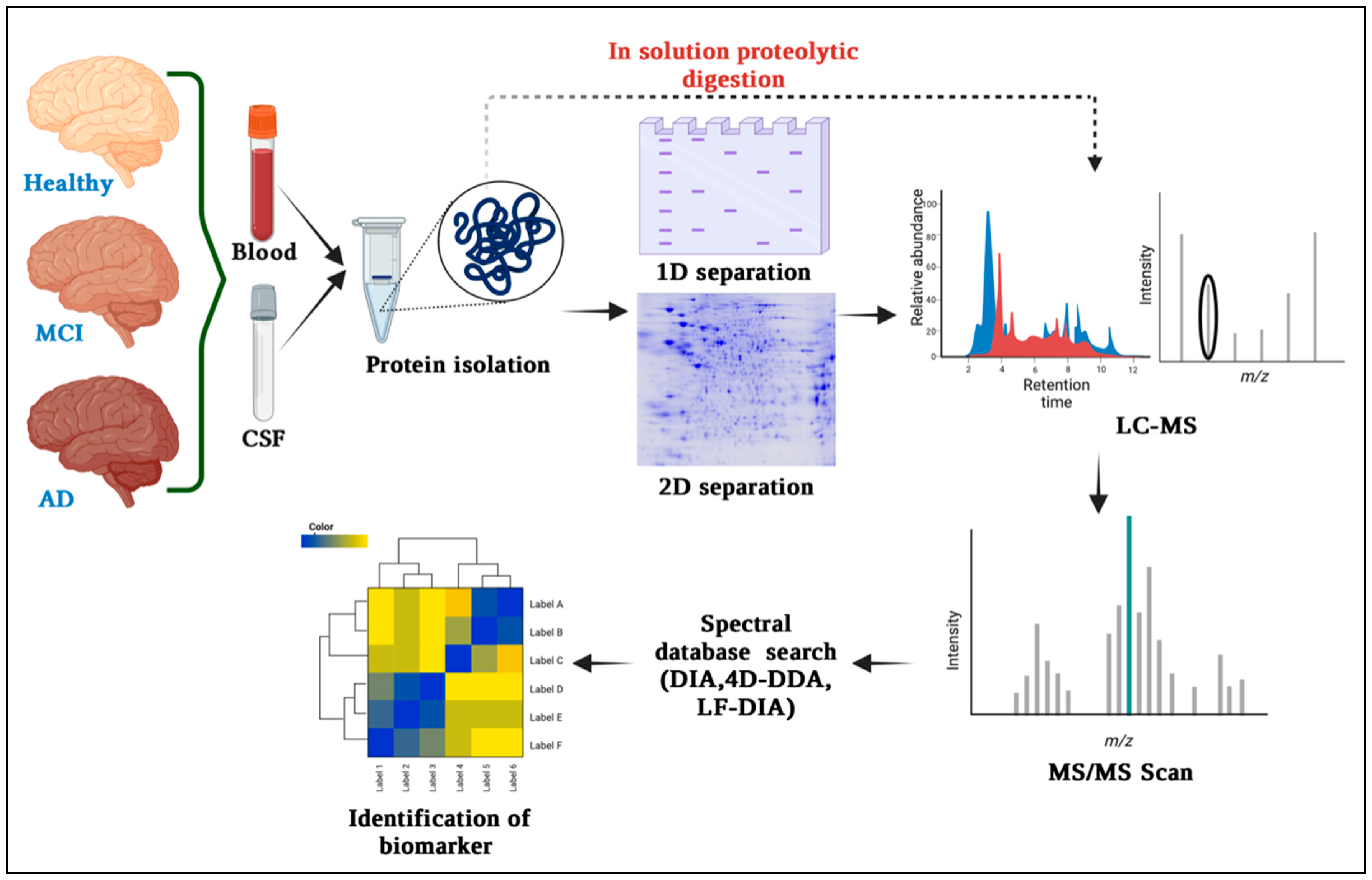
Figure 1. Overview of mass spectrometry-based proteomics techniques commonly used for biomarker discovery in Alzheimer’s disease. It includes protein separation, peptide identification, and quantification steps, which contribute to the identification of potential biomarkers.
3. Protein Microarrays and Antibody-Based Techniques
Protein microarrays are a miniaturized version of traditional biochemical assays, where hundreds to thousands of different proteins or peptides are immobilize in a spatially addressable manner on a solid substrate, such as a glass slide or a silicon chip [60][24]. These immobilized proteins can be used to study various aspects of protein function, including protein–protein interactions, protein–ligand interactions, protein-DNA interactions, enzyme activities, and post-translational modifications. In a specific investigation, scientists employed cDNA microarrays containing 18,000 genes to scrutinize cDNA samples from hippocampal CA1 neurons. These samples were obtained from Alzheimer’s patients with and without neurofibrillary tangles, along with control subjects. Similarly, prefrontal cortex samples from individuals with schizophrenia and controls were screened using 7000-gene arrays to detect gene expression variations. This screening unveiled decreased expression of genes regulating presynaptic function. Validation of the observed gene expression changes via methods like immunohistochemistry, in situ hybridization or reverse transcription polymerase chain reaction is crucial [61][25]. Another illustrative case pertains to the utilization of microarrays to examine the transcriptional profile of brain plaques from multiple sclerosis (MS) patients, juxtaposed with control brain samples. This restudy earch effectively identified the exclusive presence of osteopontin (OPN) gene expression within MS plaques. As a result, it was proposed that this pro-inflammatory molecule is generated by infiltrating T lymphocytes, microglia, and macrophages, contributing to myelin sheath damage via an autoimmune mechanism. Furthermore, it is evident that alterations in the OPN gene might affect disease progression [62][26]. In Ho et al.’s 2005 study, 6794 human genes were screened, revealing 32 aberrantly expressed genes (25 known and 7 unknown, based on EST) in the superior temporal gyrus of moderate dementia cases compared to cognitively normal controls (>1.8-fold difference) [63][27]. These findings highlight the potential significance of these genes in early stage AD. Further research is needed to elucidate their precise role(s) in AD development and progression. In Kim JR et al.’s 2003 study, microarray experiments examined gene dysregulation in AD animal models. Introducing pathogenic mutations of APP, presenilin, and tau in mice led to AD pathologies like amyloid plaques and neurofibrillary tangles [64][28]. Expression profiling identified the downstream effects of these mutations in transgenic AD animal models. In conclusion, microarray-based techniques have emerged as powerful tools for proteome-based analysis in Alzheimer’s disease (AD). They allow for the comprehensive profiling of protein expression levels and can help identify key biomarkers associated with AD pathology; unlocking the potential of Western blot analysis, an independent technique crucial for studying protein expression. Through gel electrophoresis, it separates proteins based on size and charge, followed by antibody binding to the target protein on a membrane. Detection methods then reveal vital information about the protein’s presence and abundance, making it invaluable for AD research. Antibody selection for respective Aβ recognition motifs is a crucial part of Western blotting. Both monoclonal and polyclonal types of antibodies are used in Western blotting. Some of the major antibodies are described in Table 1 below.
Table 1.
Antibodies used in Western blot analysis to identify amyloid β-protein (Aβ) with their corresponding Aβ recognition motifs and other motifs.
| Recognition Motif | Nature of Antibody | Name of Antibody | Reference |
|---|---|---|---|
| Aβ1–16 | Monoclonal | Ab9 | [65][29] |
| Aβ1–16 | Monoclonal | 6C6 | [66][30] |
| Aβ1–17 | Monoclonal | 6E10 | [67][31] |
| Aβ17–24 | Monoclonal | 4G8 | [68][32] |
| Aβ31–40 | Monoclonal | 2G3 | [69][33] |
| Aβ1–40, C-terminal | Monoclonal | BA-27 | [70][34] |
| Aβ1–42, C-terminal | Monoclonal | BC-05 | [71][35] |
| Amyloid oligomers | Monoclonal | A8 | [72][36] |
| Amyloid oligomers | Monoclonal | A11 | [73][37] |
| Amyloid oligomers | Monoclonal | NU-4 | [74][38] |
| Amyloid fibrils | Polyclonal | OC | [75][39] |
| Anti-amyloid beta precursor protein | Monoclonal | Y188 | [76][40] |
| Anti-APP | Monoclonal | A8717 | [77][41] |
| Anti-myelin basic protein | Monoclonal | MBP | [78][42] |
| Anti-kelch-like ECH-associated protein 1 | Monoclonal | KEAP1 | [79][43] |
Pryor et al. employed the A8 monoclonal antibody, designed to target oligomers, as a substitute for the preparation of Aβ1–42. The antibody A8 displayed a range of oligomer species with sizes spanning from 16.5 to 25 kDa [80][44]. In contrast, the 6E10 antibody provided poorer resolution, displaying larger species of oligomers. These findings imply that 6E10 might exhibit a more potent response to oligomers with greater molecular weight, or alternatively, the antibodies might exhibit a preference for binding to various sizes of Aβ1–42 oligomers [81][45]. While Western blotting assists in identifying intermediate Aβ oligomers, the prevalent gel smear in numerous studies suggests its inability to precisely quantify individual oligomer sizes in this range [82,83][46][47]. Interestingly, a recent study investigated a comparison between age-matched control cases exhibiting normal cognitive status (CDR 0) and individuals with mild cognitive impairment (MCI) (CDR 0.5). The researchers observed a significant N2-fold reduction in splice variants I–III of the a-type synapsin isoform within the entorhinal cortex of MCI cases [84][48]. Interestingly, there were no significant alterations noted in splice variant II of the b-type synapsin isoform or in synaptophysin within the same EC region. Notably, the modified expressions of synapsin a-type isoforms were exclusive to the EC in cases of MCI, as no observable decreases were found in the VC of the same individuals [85][49]. These groundbreaking discoveries illuminate the importance of selectively modified gene expression in the initial detectable phase of AD dementia. They offer valuable molecular evidence.
4. Advancements in High-Throughput Proteomics Technologies
Some of the high throughput proteomics technologies such as TMT (tandem mass tag), cysteine-reactive tandem mass tag (cysTMT), OxcysDML, isobaric tags for relative and absolute quantitation (iTRAQ), quantitative thiol reactivity profiling (QTRP), and electrophilic diazene probe (DiaAlk) were used for proteomics analysis in AD and other ND. In the work proposed by Mei Chen et al., 2020, TMT (tandem mass tag) was used for proteomic study of body fluids from AD patients. This technique enables relative quantitation of proteins present in multiple samples by labeling peptides with stable isotope tags that fragment into reporter-ions upon collision-induced dissociation [86][50]. Also, redox proteomics can detect hundreds to thousands of oxidized proteins in a single experiment and this is attractive for understanding the redox status of proteins. Thus, in another work in 2014, Garcia-Santamarina et al. proposed the OxcysDML method for quantifying cysteine redox in a demonstration capacity. This technique was employed to examine the liver proteome of a mouse model with Alzheimer’s disease, with the goal of enhancing comprehension regarding redox chemistry in the condition. As per their results, nearly 90% of cysteine was observed in its reduced state within the living organism. Given the prominence of cysteine in the mouse proteome (accounting for around 14% of all in silico tryptic peptides containing cysteine), the scientists approximated that only about 2% of tryptic peptides (equivalent to roughly 4 μg per sample) underwent enrichment and were subsequently subjected to analysis using OxcysDML [87][51]. In 2008, Bronwen Martin conducted a study where they demonstrated the use of the iTRAQ control protocol alongside 3xTgAD tissue samples. Concurrently, both the control and AD samples underwent treatment throughout the labeling process. This labeling protocol encompassed multiple stages: protein reduction and cysteine blocking, protein trypsin digestion, peptide labeling using iTRAQ reagents, merging the samples for comparison, employing strong cation exchange chromatography, conducting solid phase extraction for desalination, and concluding with LC/MS/MS analysis [88][52]. Through this comprehensive approach, they were able to unravel some of the intricate proteome changes that occur in a mouse model of Alzheimer’s disease. These discoveries hold the promise of unveiling fresh therapeutic avenues for Alzheimer’s disease (AD) and other neurodegenerative disorders (ND). Progress in high-throughput proteomics technologies has transformed the realm of proteomics, empowering scientists to examine extensive protein samples rapidly and efficiently, marking a revolutionary shift in the field. Through innovative techniques, such as mass spectrometry-based approaches and quantitative proteomics, scientists can now gain deeper insights into complex biological systems, paving the way for groundbreaking discoveries in disease mechanisms, drug development, and personalized medicine [89][53]. As these technologies continue to evolve, the future holds great promise for unraveling the intricacies of the proteome and its vital role in various physiological and pathological processes.
References
- Garfin, D.E. One-dimensional gel electrophoresis. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 1990; Volume 182, pp. 425–441.
- Lee, P.Y.; Saraygord-Afshari, N.; Low, T.Y. The evolution of two-dimensional gel electrophoresis-from proteomics to emerging alternative applications. J. Chromatogr. A 2020, 1615, 460763.
- Shevchenko, G.; Konzer, A.; Musunuri, S.; Bergquist, J. Neuroproteomics tools in clinical practice. Biochim. Biophys. Acta (BBA)—Proteins Proteom. 2015, 1854, 705–717.
- Friedman, D.B.; Hoving, S.; Westermeier, R. Isoelectric focusing and two-dimensional gel electrophoresis. Methods Enzymol. 2009, 463, 515–540.
- Santoni, V.; Molloy, M.; Rabilloud, T. Membrane proteins and proteomics: Un amour impossible? Electrophor. Int. J. 2000, 21, 1054–1070.
- Rabilloud, T. Use of thiourea to increase the solubility of membrane proteins in two-dimensional electrophoresis. Electrophoresis 1998, 19, 758–760.
- Yan, J.X.; Kett, W.C.; Herbert, B.R.; Gooley, A.A.; Packer, N.H.; Williams, K.L. Identification and quantitation of cysteine in proteins separated by gel electrophoresis. J. Chromatogr. A 1998, 813, 187–200.
- Görg, A.; Klaus, A.; Lück, C.; Weiland, F.; Weiss, W. Two-dimensional electrophoresis with immobilized pH gradients for proteome analysis. A Lab. Man. 2007, 1855, 125–129.
- Craft, G.E.; Chen, A.; Nairn, A.C. Recent advances in quantitative neuroproteomics. Methods 2013, 61, 186–218.
- Lull, M.E.; Erwin, M.S.; Morgan, D.; Roberts, D.C.; Vrana, K.E.; Freeman, W.M. Persistent proteomic alterations in the medial prefrontal cortex with abstinence from cocaine self-administration. Proteom. Clin. Appl. 2009, 3, 462.
- Naseri, N.N.; Ergel, B.; Kharel, P.; Na, Y.; Huang, Q.; Huang, R.; Dolzhanskaya, N.; Burré, J.; Velinov, M.T.; Sharma, M. Aggregation of mutant cysteine string protein-α via Fe–S cluster binding is mitigated by iron chelators. Nat. Struct. Mol. Biol. 2020, 27, 192–201.
- Freeman, W.M.; Hemby, S.E. Proteomics for protein expression profiling in neuroscience. Neurochem. Res. 2004, 29, 1065–1081.
- Baggerman, G.; Vierstraete, E.; De Loof, A.; Schoofs, L. Gel-based versus gel-free proteomics: A review. Comb. Chem. High Throughput Screen. 2005, 8, 669–677.
- Thiede, B.; Höhenwarter, W.; Krah, A.; Mattow, J.; Schmid, M.; Schmidt, F.; Jungblut, P.R. Peptide mass fingerprinting. Methods 2005, 35, 237–247.
- Háda, V.; Bagdi, A.; Bihari, Z.; Timári, S.B.; Fizil, Á.; Szántay, C., Jr. Recent advancements, challenges, and practical considerations in the mass spectrometry-based analytics of protein biotherapeutics: A viewpoint from the biosimilar industry. J. Pharm. Biomed. Anal. 2018, 161, 214–238.
- Stump, M.J.; Fleming, R.C.; Gong, W.H.; Jaber, A.J.; Jones, J.J.; Surber, C.W.; Wilkins, C.L. Matrix-assisted laser desorption mass spectrometry. Appl. Spectrosc. Rev. 2002, 37, 275–303.
- Domon, B.; Aebersold, R. Mass spectrometry and protein analysis. Science 2006, 312, 212–217.
- Blackstock, W.P.; Weir, M.P. Proteomics: Quantitative and physical mapping of cellular proteins. Trends Biotechnol. 1999, 17, 121–127.
- Hernandez, P.; Müller, M.; Appel, R.D. Automated protein identification by tandem mass spectrometry: Issues and strategies. Mass Spectrom. Rev. 2006, 25, 235–254.
- Villar-Garea, A.; Griese, M.; Imhof, A. Biomarker discovery from body fluids using mass spectrometry. J. Chromatogr. 2007, 849, 105–114.
- Palubeckaitė, I. Analysis of Three-Dimensional Cell Cultures Using Mass Spectrometry Imaging; Sheffield Hallam University: Sheffield, UK, 2018.
- Grela, A.; Turek, A.; Piekoszewski, W. Application of matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS) in Alzheimer’s disease. Clin. Chem. Lab. Med. 2012, 50, 1297–1304.
- Korolainen, M. Proteomic Analysis of Post-Translationally Modified Proteins in Alzheimer’s Disease (Alzheimerin Taudin Post-Translationaalisesti Muunneltujen Proteiinien Kartoittaminen Proteomiikan Avulla); Kuopion Yliopisto: Kuopio, Finland, 2006.
- Sobek, J.; Bartscherer, K.; Jacob, A.; Hoheisel, J.D.; Angenendt, P. Microarray technology as a universal tool for high-throughput analysis of biological systems. Comb. Chem. High Throughput Screen. 2006, 9, 365–380.
- Ginsberg, S.D.; Hemby, S.E.; Lee, V.M.; Eberwine, J.H.; Trojanowski, J.Q. Expression profile of transcripts in Alzheimer’s disease tangle-bearing CA1 neurons. Ann. Neurol. 2000, 48, 77–87.
- Housley, W.J.; Pitt, D.; Hafler, D.A. Biomarkers in multiple sclerosis. Clin. Immunol. 2015, 161, 51–58.
- Ho, L.; Fivecoat, H.; Wang, J.; Pasinetti, G.M. Alzheimer’s disease biomarker discovery in symptomatic and asymptomatic patients: Experimental approaches and future clinical applications. Exp. Gerontol. 2010, 45, 15–22.
- Jankowsky, J.L.; Zheng, H. Practical considerations for choosing a mouse model of Alzheimer’s disease. Mol. Neurodegener. 2017, 12, 1–22.
- Levites, Y.; Das, P.; Price, R.W.; Rochette, M.J.; Kostura, L.A.; McGowan, E.M.; Murphy, M.P.; Golde, T.E. Anti-Aβ42- and anti-Aβ40-specific mAbs attenuate amyloid deposition in an Alzheimer disease mouse model. J. Clin. Investig. 2006, 116, 193–201.
- Seubert, P.; Oltersdorf, T.; Lee, M.G.; Barbour, R.; Blomquist, C.; Davis, D.L.; Bryant, K.; Fritz, L.C.; Galasko, D.; Thal, L.J.; et al. Secretion of β-amyloid precursor protein cleaved at the amino terminus of the β-amyloid peptide. Nature 1993, 361, 260–263.
- Deng, J.; Hou, H.; Giunta, B.; Mori, T.; Wang, Y.J.; Fernandez, F.; Weggen, S.; Araki, W.; Obregon, D.; Tan, J. Autoreactive-Aβ antibodies promote APP β-secretase processing. J. Neurochem. 2012, 120, 732–740.
- Baghallab, I.; Reyes-Ruiz, J.M.; Abulnaja, K.; Huwait, E.; Glabe, C. Epitomic characterization of the specificity of the anti-amyloid Aβ monoclonal antibodies 6E10 and 4G8. J. Alzheimer’s Dis. 2018, 66, 1235–1244.
- Walsh, D.M.; Hartley, D.M.; Condron, M.M.; Selkoe, D.J.; Teplow, D.B. In vitro studies of amyloid β-protein fibril assembly and toxicity provide clues to the aetiology of Flemish variant (Ala692→ Gly) Alzheimer’s disease. Biochem. J. 2001, 355, 869–877.
- Borchelt, D.R.; Thinakaran, G.; Eckman, C.B.; Lee, M.K.; Davenport, F.; Ratovitsky, T.; Prada, C.M.; Kim, G.; Seekins, S.; Yager, D.; et al. Familial Alzheimer’s disease–linked presenilin 1 variants elevate Aβ1–42/1–40 ratio in vitro and in vivo. Neuron 1996, 17, 1005–1013.
- Kawarabayashi, T.; Younkin, L.H.; Saido, T.C.; Shoji, M.; Ashe, K.H.; Younkin, S.G. Age-dependent changes in brain, CSF, and plasma amyloid β protein in the Tg2576 transgenic mouse model of Alzheimer’s disease. J. Neurosci. 2001, 21, 372–381.
- Zhang, Y.; He, J.S.; Wang, X.; Wang, J.; Bao, F.X.; Pang, S.Y.; Yin, F.; Hu, H.G.; Peng, X.L.; Sun, W.M.; et al. Administration of Amyloid-β 42 Oligomer-Specific Monoclonal Antibody Improved Memory Performance in SAMP8 Mice. J. Alzheimer’s Dis. 2011, 23, 551–561.
- Kayed, R.; Canto, I.; Breydo, L.; Rasool, S.; Lukacsovich, T.; Wu, J.; Albay, R.; Pensalfini, A.; Yeung, S.; Head, E.; et al. Conformation dependent monoclonal antibodies distinguish different replicating strains or conformers of prefibrillar Aβ oligomers. Mol. Neurodegener. 2010, 5, 57.
- Lambert, M.P.; Velasco, P.T.; Chang, L.; Viola, K.L.; Fernandez, S.; Lacor, P.N.; Khuon, D.; Gong, Y.; Bigio, E.H.; Shaw, P.; et al. Monoclonal antibodies that target pathological assemblies of Aβ. J. Neurochem. 2007, 100, 23–35.
- Isas, J.M.; Luibl, V.; Johnson, L.V.; Kayed, R.; Wetzel, R.; Glabe, C.G.; Langen, R.; Chen, J. Soluble and mature amyloid fibrils in drusen deposits. Investig. Ophthalmol. Vis. Sci. 2010, 51, 1304–1310.
- Chebli, J.; Rahmati, M.; Lashley, T.; Edeman, B.; Oldfors, A.; Zetterberg, H.; Abramsson, A. The localization of amyloid precursor protein to ependymal cilia in vertebrates and its role in ciliogenesis and brain development in zebrafish. Sci. Rep. 2021, 11, 19115.
- García-Ayllón, M.S.; Lopez-Font, I.; Boix, C.P.; Fortea, J.; Sánchez-Valle, R.; Lleó, A.; Molinuevo, J.L.; Zetterberg, H.; Blennow, K.; Sáez-Valero, J. C-terminal fragments of the amyloid precursor protein in cerebrospinal fluid as potential biomarkers for Alzheimer disease. Sci. Rep. 2017, 7, 2477.
- Jingwu, Z.; Vandenbark, A.A.; Jacobs, M.P.; Offner, H.; Raus, J.C. Murine monoclonal anti-myelin basic protein (MBP) antibodies inhibit proliferation and cytotoxicity of MBP-specific human T cell clones. J. Neuroimmunol. 1989, 24, 87–94.
- Kang, C.; Jeong, S.; Kim, J.; Ju, S.; Im, E.; Heo, G.; Park, S.; Yoo, J.W.; Lee, J.; Yoon, I.S.; et al. N-Acetylserotonin is an oxidation-responsive activator of Nrf2 ameliorating colitis in rats. J. Pineal Res. 2023, 74, e12835.
- Pryor, N.E.; Moss, M.A.; Hestekin, C.N. Unraveling the early events of amyloid-β protein (Aβ) aggregation: Techniques for the determination of Aβ aggregate size. Int. J. Mol. Sci. 2012, 13, 3038–3072.
- Puig Gomà-Camps, E. Structural Characterization of Amyloid Beta Oligomers with Functional Links Associated to Alzheimer’s Disease. 2019. Available online: https://dialnet.unirioja.es/servlet/tesis?codigo=250722 (accessed on 29 August 2023).
- Miyoshi, E.; Bilousova, T.; Melnik, M.; Fakhrutdinov, D.; Poon, W.W.; Vinters, H.V.; Miller, C.A.; Corrada, M.; Kawas, C.; Bohannan, R.; et al. Exosomal tau with seeding activity is released from Alzheimer’s disease synapses, and seeding potential is associated with amyloid beta. Lab. Investig. 2021, 101, 1605–1617.
- Ying, Z.; Xin, W.; Jin-Sheng, H.; Fu-Xiang, B.; Wei-Min, S.; Xin-Xian, D.; Xiao-Bo, W.; Yi-Qin, L.; Xian-Xian, Z.; Hong-Gang, H.; et al. Preparation and characterization of a monoclonal antibody with high affinity for soluble Aβ oligomers. Hybridoma 2009, 28, 349–354.
- Vaillant-Beuchot, L.; Mary, A.; Pardossi-Piquard, R.; Bourgeois, A.; Lauritzen, I.; Eysert, F.; Kinoshita, P.F.; Cazareth, J.; Badot, C.; Fragaki, K.; et al. Accumulation of amyloid precursor protein C-terminal fragments triggers mitochondrial structure, function, and mitophagy defects in Alzheimer’s disease models and human brains. Acta Neuropathol. 2021, 141, 39–65.
- Yang, Y.W.; Hsu, K.C.; Wei, C.Y.; Tzeng, R.C.; Chiu, P.Y. Operational Determination of Subjective Cognitive Decline, Mild Cognitive Impairment, and Dementia Using Sum of Boxes of the Clinical Dementia Rating Scale. Front. Aging Neurosci. 2021, 13, 705782.
- Pham, T.K.; Buczek, W.A.; Mead, R.J.; Shaw, P.J.; Collins, M.O. Proteomic approaches to study cysteine oxidation: Applications in neurodegenerative diseases. Front. Mol. Neurosci. 2021, 14, 678837.
- García-Santamarina, S.; Boronat, S.; Domènech, A.; Ayté, J.; Molina, H.; Hidalgo, E. Monitoring in vivo reversible cysteine oxidation in proteins using ICAT and mass spectrometry. Nat. Protoc. 2014, 9, 1131–1145.
- Martin, B.; Brenneman, R.; Becker, K.G.; Gucek, M.; Cole, R.N.; Maudsley, S. iTRAQ analysis of complex proteome alterations in 3xTgAD Alzheimer’s mice: Understanding the interface between physiology and disease. PLoS ONE 2008, 3, e2750.
- Lundberg, E.; Borner, G.H. Spatial proteomics: A powerful discovery tool for cell biology. Nat. Rev. Mol. Cell Biol. 2019, 20, 285–302.
More