1. Introduction
Traditional strategies in the treatment of cancer include surgical resection, radiation, chemotherapy, or some combination thereof. Surgery is not an option for many cancers, and in those cases where it is possible, success relies on both the ability of the patient to survive the surgery and the ability of the surgeon to completely remove all of the diseased tissues
[1]. Radiation and chemotherapy serve to eradicate malignant cells; however, these treatments are not selective, causing off-target toxicity in healthy tissues that leads to significant undesired side effects and the potential for highly dangerous immunosuppression
[2]. In addition, in cases where chemotherapy fails to completely eradicate the cancer, there is significant risk for the development of resistance to the treatment, increasing the risk of both metastasis and disease relapse
[3].
Cancer immunotherapy, which has attracted considerable attention in recent years, aims to sensitize the patient’s immune system to the disease for its selective eradication
[4]. Various immunotherapy strategies continue to be explored, with several having gained clinical approval
[5]. Most prevalent among the clinically approved immunotherapies are checkpoint inhibitors (ICIs), which modulate the aberrant regulation that prevents the immune system from attacking cancer cells
[6]. Unfortunately, the benefits of ICIs are limited to specific cohorts of patients who have inherently higher T cell levels at the tumor site
[7]. ICIs also suffer from other limitations, including the development of treatment resistance, and toxicity that arises from the disruption of the natural homeostatic balance of the immune system
[8]. Ultimately hindering the success of any cancer immunotherapy is the tumor microenvironment (TME), which dynamically regulates therapeutic responses and contributes to treatment resistance
[9]. Large populations of immunosuppressive cells, including tumor-associated macrophages (TAMs) and regulatory T cells (Tregs), contribute to a non-inflamed “cold” TME, which limits the infiltration and activity of T cells within the tumor
[10]. In addition, down-regulation of major histocompatibility complex class 1 (MHC-1) immune ligand expression on the surface of cancer cells significantly hampers the potential for immunosurveillance
[11][12][11,12]. Thus, there is a considerable need for novel immunotherapy strategies that can induce an inflamed “hot” TME that promotes cytotoxic T lymphocyte (CTL) and natural killer (NK) cell responses by way of balancing TME immunosuppressive effects, increasing tumor immunogenicity via enhanced tumor-associated antigen presentation, and increasing the trafficking and infiltration of T cells into the tumor
[13][14][13,14].
The stimulator of interferon genes (STING) cellular signaling pathway is emerging as a promising immunotherapeutic target to combat the immunosuppressive TME
[15]. The STING pathway is an important player in the DNA damage response (DDR) and is involved in monitoring the cytosol of cells for the presence of foreign or damaged DNA, a distress signal that arises from a multitude of potential threats
[16]. The STING protein is activated by the cyclic dinucleotide (CDN) 2′3′-cyclic guanosine monophosphate–adenosine monophosphate (cGAMP), a second messenger molecule that is produced by cGAMP synthase (cGAS) after it detects the presence of cytosolic double-stranded DNA (dsDNA)
[17][18][17,18]. The recognition of dsDNA by cGAS is mostly sequence-independent, with the minimal length for cGAS activation being 20–40 base pairs and varying between species
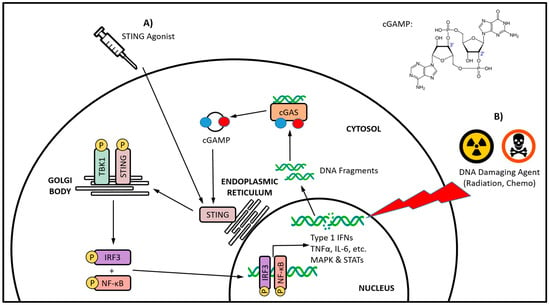
[19][20][19,20]. Upon binding cGAMP, the STING homodimer undergoes extensive conformational rearrangements and oligomerization, and ultimately translocates from the endoplasmic reticulum to the Golgi body (
Figure 1)
[21][22][23][24][21,22,23,24]. The recruitment and activation of TANK-binding kinase 1 (TBK1) by autophosphorylation leads to the phosphorylation of STING
[25][26][25,26]. The resulting STING–TBK1 complex phosphorylates interferon regulatory factor 3 (IRF3), leading to its homodimerization and nuclear translocation, where it induces target gene expression
[27][28][27,28]. The downstream effects of STING pathway activation are variable, and are dependent on the particular cell type, as well as the intensity and duration of activation
[29]. However, a characteristic of STING pathway signaling is the secretion of type I IFNs
[30][31][32][30,31,32]. In some settings, STING signaling can also be associated with the activation of nuclear factor κB (NFκB), mitogen-activated protein (MAP) kinases, and signal transducer and activator of transcription (STAT) transcription factors
[33][34][35][36][37][33,34,35,36,37].
Figure 1. The cGAS-STING axis signaling and a comparison of direct versus indirect STING activation. Direct STING activation (A) is being targeted with STING agonists, whereas indirect activation (B) occurs via cGAS sensing of cytosolic DNA fragments, and its subsequent endogenous synthesis of the canonical CDN STING agonist cGAMP. The DNA damage induced by traditional cancer therapies, such as radiation and certain chemotherapeutics, has been increasingly linked with indirect STING activation. Whether directly or indirectly activated, the translocation of STING from the endoplasmic reticulum to the Golgi body initiates a series of phosphorylation-based signaling events culminating in the activation of the IRF3 and NF-κB transcription factors, and subsequent production of type I IFNs and various other proinflammatory cytokines.
Type I IFNs directly regulate the transcription of over 100 genes that influence many key aspects of cell survival and immunity
[38]. In the cancer setting, type I IFNs have been shown to directly inhibit the proliferation of tumor cells
[39][40][41][39,40,41], and disrupt tumor vasculature
[42][43][42,43]. While the production of type I IFNs is a critical component of STING signaling for promoting antitumor immunity, the other IFN-independent signaling pathways downstream of STING activation also play a key role in cancer immune regulation
[44]. Taken together, the cancer immunotherapeutic potential of STING signaling arises from its capacity to promote a wide array of antitumor immune responses (
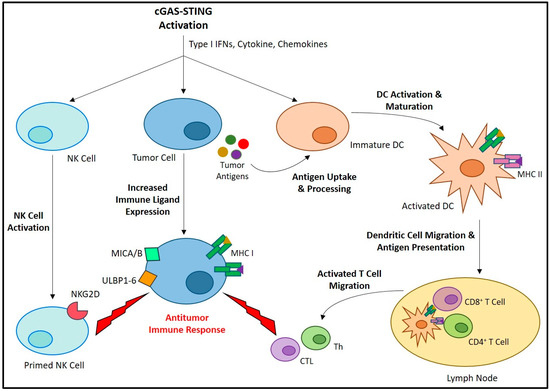
Figure 2)
[45]. STING activation promotes the maturation of professional antigen-presenting cells (APCs), leading them to express various costimulatory molecules and pro-inflammatory cytokines required for T cell priming and activation
[46][47][48][49][46,47,48,49]. Both T helper lymphocyte (Th)
[50][51][50,51] and CTL
[52][53][52,53] responses are known to be enhanced by STING-induced APC maturation
[54][55][54,55]. Furthermore, a balanced type 1/type 2 (Th1/Th2), or even Th1 biased phenotype
[56] have been reported in response to STING-induced IFN signaling, thereby promoting M1 macrophage polarization
[57][58][57,58]. Compared to M2 macrophages, which are known to be immunosuppressive, M1 macrophages are more supportive of antitumor immune responses
[59][60][61][59,60,61]. STING-induced pro-inflammatory cytokines, including interleukin 6 (IL-6) and tumor necrosis factor alpha (TNF-α), as well as reactive nitrogen and oxygen species (ROS), can promote M1 macrophage polarization and the repolarization of immunosuppressive TAMs
[59][62][63][59,62,63]. STING signaling also generates a chemokine gradient, including CXCL10, CCL5, and CXCL9, that can guide the recruitment and activation of T cells
[52][64][65][52,64,65] and NK cells
[66] within the tumor. STING signaling promotes MHC-1 expression on the cancer cell surface that is required for CTL recognition and eradication
[67]. Furthermore, STING signaling has been shown to upregulate expression of the ligands of NK group 2, member D (NKG2D), an NK cell-specific immunoreceptor necessary for the recognition and elimination of cancer cells
[68].
Figure 2. The role of the cGAS-STING axis in promoting anticancer immunity. The activation of cGAS-STING and the resulting type I IFNs, cytokines, and chemokines induce a variety of cell-specific responses that all contribute to antitumor immune activity. Activation of immature DCs leads to increased antigen uptake/processing, maturation, and lymph node migration. Here, the DCs present antigens to naïve T cells, and potentiate their differentiation into Th and CTL, which migrate and infiltrate the tumor site where they carry out antitumor responses. A similar process occurs in NK cells, in which their priming leads to an increased expression of immune receptors, including NKG2D. These cellular immune responses are promoted by requisite increases in tumor cell immune ligand expression, including MHC-1 for CTL recognition, as well as MHC class 1 chain-related protein A and B (MICA/B) and UL16-binding proteins 1-6 (ULBP1-6) for recognition by NKG2D.
The promising immunostimulatory potential of the STING pathway has fueled considerable efforts geared towards the development of STING agonist therapeutics (
Figure 1A) (reviewed in
[69]). STING agonists have shown success for generating antitumor immunity against a wide range of cancer types in preclinical research, prompting numerous clinical trials (reviewed in
[15]). However, both the safety and efficacy of STING agonists are limited by numerous pharmacological and drug delivery challenges, including metabolic stability issues, low cellular uptake/intracellular delivery, and the potential for immune-related adverse events including cytotoxicity
[70][71][70,71]. Another major concern in the development of STING agonists is systemic administration, and the lack of tissue or cell specificity. In addition to cancer cells, STING is expressed in many other cell types that may have adverse responses impacting therapeutic outcomes
[72]. While STING activation in extratumoral cell populations may result in antitumor effects, the ideal location for STING activation is within the tumor site, where it can generate the necessary local immunostimulatory effects
[73]. Restricting systemic-wide STING activation is necessary to minimize nonspecific systemic inflammatory responses
[74]. Ultimately, while STING pathway agonists offer considerable promise for cancer immunotherapy, none have yet gained clinical approval.
Despite the promising immunotherapeutic potential of the STING pathway for cancer treatment, there are several critical and inherent concerns. The duration of STING pathway stimulation is an important consideration, as it can drastically influence the balance of immunological outcomes. Localized and acute activation of the STING pathway supports an appropriate level of immune activation for cancer eradication. However, chronic STING signaling is implicated in a variety of inflammation-driven diseases
[75]. In addition, prolonged STING pathway activation can also lead to cancer development and metastasis
[76][77][78][79][76,77,78,79]. While the production of type I IFNs is a critical component of STING-signaling-induced antitumor immunity, recent studies have suggested that type I IFNs may actually impair anticancer immunity and cause treatment resistance. For example, IFN-β has been shown to increase the levels of programmed cell death ligands 1 (PD-L1) and 2 (PD-L2), which are known to contribute to immune escape by cancer cells
[80][81][80,81]. Type I IFNs have also been shown to contribute to unexpected immune toxicity during cancer immunotherapy
[82]. In addition, the other IFN-independent signaling pathways downstream of STING activation also play a key role in immune regulation, and can contribute to tumor immune evasion
[44][83][44,83].
It has been known for some time that traditional cancer treatments such as radiation and chemotherapy have beneficial immunostimulatory effects that continue to be reported
[84]. These cancer treatments stimulate aspects of both the innate and adaptive immune systems via several different mechanisms. The cytotoxic activity of radiation and chemotherapy have been linked to the induction of immunogenic cell death, which leads to the release of specific signals that trigger the phagocytosis of cellular debris and the maturation of APC
[85]. In addition, these traditional cancer treatments have been reported to alleviate tumor-induced immunosuppressive mechanisms
[86][87][88][86,87,88], and cause transient lymphodepletion that allows for localized immune cell replenishment
[89]. Mechanistically, common to the majority of these traditional cancer treatments is their DNA-damaging activity, which ultimately leads to their cytotoxicity. Opposing this activity is the DDR, which is a highly organized network of interconnected components that are responsible for the repair of damaged DNA and the maintenance of genomic stability
[90]. Defects in the DDR lead to an imbalance between DNA damage and repair that can drive tumorigenesis, inflammatory cytokine secretion, and aberrant immune responses
[91]. Emerging evidence is outlining the critical link between the DDR and antitumor immunity, in that it shapes the innate immune response and how the adaptive immune system is recruited to tumors
[92]. Most immune-related DDR components and immune responses converge upon the STING-IFN signaling pathway
[93]. It is not surprising then, that many of the traditional, DNA-damaging cancer treatments are being linked with indirect and iatrogenic STING activation (
Figure 1B). However, the significance of said STING activation in cancer patients and potential immunotherapeutic applications are currently unclear.
2. Conventional Cancer Therapies and cGAS-STING Activation
2.1. Antimitotic Agents
Microtubule-targeting agents (MTAs), such as Taxol, have been used extensively for the treatment of rapidly dividing cancers, and cause cell death by inducing mitotic arrest and eventual apoptosis
[94][121]. While MTAs do not directly interact with DNA or members of the DDR, their activity has been linked with the induction of limited degrees of DNA damage resulting from the partial activation of apoptosis and its associated nucleases
[95][96][122,123]. MTAs are also known to disrupt the intracellular trafficking of various DNA repair proteins, thus augmenting the toxicity of DNA-damaging chemotherapies which is the basis for their combination as a common anticancer regimen
[97][124].
Zierhut et al. demonstrated that Taxol-induced mitotic arrest in the HeLa cervical cancer cell line resulted in a slow accumulation of IRF3 phosphorylation in a cGAS-dependent manner
[98][94]. This triggered apoptosis via the transcription-independent release of BCL-xL-dependent suppression of mitochondrial outer membrane permeabilization. The authors went on to demonstrate that histone-wrapped nucleosomic DNA had a higher affinity for cGAS than naked DNA, and prevented cytoplasmic chromosomal DNA from activating cGAS during normal mitosis by competitive inhibition. In addition, the cGAS expression level was shown to correlate with Taxol sensitivity in a panel of breast cancer cell lines, and promoted the response to Taxol in a mouse xenograft model of cervical cancer. Lohard et al. showed that Taxol induced DNA micronuclei in breast cancer cell lines and patient-derived mouse xenografts that led to the downstream activation of the cGAS-STING pathway
[99][95]. The resulting secretion of type I IFN and TNF-α formed a proapoptotic paracrine secretome, which triggered NOXA expression in neighboring cells, and increased their sensitivity to the inhibition of BCL-xL. Hu et al. demonstrated that cytotoxic doses of Taxol induced cGAS-positive micronucleation and subsequent STING signaling in various triple-negative breast cancer (TNBC) cell lines
[100][96]. The resulting cytokines and chemokines from this response were able to induce a M1-like polarization of THP-1 derived macrophages in vitro. The authors further showed that similar doses of other MTAs, including vinorelbine and eribulin, also induced cGAS-positive micronucleation; however, evidence of subsequent downstream STING signaling was not provided. In a somewhat contradictory report to that of Hu et al., Fermaintt et al. showed that cytotoxic doses of eribulin, but not Taxol, induced cGAS-STING-depended expression of type I IFNs in both myeloid and TNBC cells in vitro
[101][97]. The cGAS-STING pathway activation by eribulin was further demonstrated to be mediated by the accumulation of cytoplasmic mitochondrial DNA. Overall, while these results suggest there may be the potential that MTAs, in general, may be able to activate the cGAS-STING signaling pathway via the DNA damage they induce, further work will need to confirm this.
2.2. DDR Inhibitors
2.2.1. Topoisomerase Inhibitors
Topoisomerases are enzymes that catalyze changes in the intertwined double-helical structure of DNA
[102][125]. They are critical for transcription, replication, and DNA repair
[102][103][125,126]. There are two classes of topoisomerases, which differ in the mechanism by which they facilitate the transient breakage of DNA strands. Topoisomerase I (Topo I) catalyzes changes in DNA structure via single-strand breaks (SSB), whereas topoisomerase II (Topo II) functions through double-strand breaks (DSB)
[104][127]. Topoisomerase inhibitors, such as camptothecin, topotecan, doxorubicin, and daunorubicin, are used as both antibacterials and chemotherapeutics, where they interfere with the catalytic cycle of the enzyme and lead to elevated levels of covalent enzyme–DNA complexes
[105][128]. Ultimately, irreversible DNA damage results in having highly prevalent DSB occurring upon collision of the replicative machinery with drug-stabilized topoisomerase–DNA complexes
[106][129]. Lymphodepletion is one of the most common side effects associated with topoisomerase inhibitors, and may result in detrimental immunosuppression
[107][130].
Pépin et al. showed that low doses of the topo I inhibitor camptothecin induced minor levels of DNA damage that resulted in a strong cGAS-STING-dependent antiviral gene response in viral oncogene-expressing mouse embryonic fibroblast cells
[108][98]. The viral oncogenes were further shown to potentiate the leakage of damaged DNA into the cytoplasm, which was critical for subsequent cGAS recruitment. As part of a chemoimmunotherapy strategy, Cao et al. developed a hybrid prodrug made up of both camptothecin and the alkylating agent cisplatin to specifically promote DNA damage-associated STING activation in tumor cells
[109][99]. Encompassing the chemotherapy hybrid was a ROS-sensitive polymeric nanoparticle, which improved the delivery, DNA-damaging activity, and subsequent cGAS-STING pathway activation in several cancer cell lines in vitro. In addition, the system activated T lymphocytes, promoted CD8+ T cell transformation into memory cells, and induced strong adaptive antitumor immune responses in an in vivo murine colorectal cancer model. The authors did not demonstrate the degree to which degree each DNA-damaging agent contributed to the overall cGAS-STING-driven response. Working with a camptothecin derivative (7-ethyl-10-hydroxycamptothecis, SN38), Zhao et al. demonstrated that the compound induced DNA damage in tumor cells which resulted in the passage of DNA-containing exosomes to APCs and subsequent cGAS-STING pathway activation
[110][100]. The SN38 was incorporated into a polymeric conjugate capable of self-assembly into nanoparticles, which not only reduced the toxicity of the drug, but also improved its ability to activate the cGAS-STING pathway and induce strong antitumor immune responses in an in vivo murine model of breast cancer. Another example of a camptothecin derivative is topotecan, which was shown by Kitai et al. to trigger STING-dependent DC activation and cytokine release
[111][101]. Various cancer cell lines treated with cytotoxic doses of the drug released DNA-containing exosomes that were taken up by DCs in a paracrine fashion. In an in vivo mouse model of breast cancer, topotecan induced significant antitumor immune responses that were driven by the infiltration of activated DCs and CD8+ T cells.
Luthra et al. demonstrated that non-cytotoxic doses of the topo II inhibitors doxorubicin and daunorubicin triggered a cGAS-STING-dependent IFN induction in HEK 293T and A549 cells expressing the Ebola virus VP35 protein
[112][102]. The response induced by these compounds was further shown to inhibit Ebola virus replication in vitro. With cytotoxic doses of the topo II inhibitor etoposide, Wang et al. demonstrated an induction of DNA damage that resulted in both STING-dependent type I IFN signaling, and NF-κB activation in various cancer cell lines in vitro
[113][103]. They went on to demonstrate activated antitumor T cell responses, both in vitro and in vivo, which potentiated the antitumor efficacy of anti-PD1 antibody treatments in several different mouse tumor models. Interestingly, Dunphy et al. demonstrated that non-cytotoxic dose of etoposide induced an early non-canonical activation of STING in HaCaT keratinocytes that was independent of cGAS
[114][104]. No production of the cGAMP second messenger, nor STING phosphorylation or translocation from the ER were observed. The DNA repair proteins ataxia telangiectasia mutated (ATM) and poly(ADP-ribose) polymerase 1 (PARP1) were required for the induction of this innate immune response, together with the DNA binding protein gamma-interferon-inducible protein 16 (IFI16), p54, and the E3 ubiquitin ligase TRAF6. The resulting signaling complex yielded an alternative STING-dependent expression program predominantly skewed towards NFκB signaling, with only a minor contribution from IRF3.
2.2.2. PARP Inhibitors
The PARP family of proteins are involved in a variety of cellular processes that include chromatin remodeling, as well as the transcription, replication, and repair of DNA damage
[115][131]. The main role of PARP in the DDR is the recruitment of other DNA-repairing enzymes via the synthesis of a polymeric ADP-ribose chain signal after detecting DNA damage
[116][132]. PARPs are involved in the base-excision repair of DNA SSB and the resection of DSB via both HR and non-homologous end joining mechanisms
[117][118][133,134]. The DNA repair activity of certain PARP proteins is exploited for survival by certain cancers that are defective in homologous repair (HR) mechanisms, and are therefore sensitive to its inhibition
[119][135]. Several PARP inhibitors (PARPi), such as olaparib, rucaparib, and talazoparib, have been approved as targeted chemotherapeutics for the treatment of cancers with mutations in the essential HR genes breast-cancer-associated 1 and 2 (BRCA1 and BRCA2), where the resulting accumulation of DNA DSB leads to genomic instability and eventual cell death
[120][136]. While PARP-inhibitor-induced DNA damage is known to have beneficial immunostimulatory effects, which include the activation of the cGAS-STING pathway as discussed below, PARP inhibitors also upregulate immunosuppressive PD-L1
[116][132].
Chabanon et al. demonstrated that cytotoxic doses of the PARPis olaparib and rucaparib induced DNA damage and cGAS-STING-dependent type I IFN signaling in both a BRCA1-deficient TNBC cell line, and lung cancer cells lacking the excision repair cross-complementation groups 1 (ERCC1) protein, a common DDR defect in non-small-cell lung cancers (NSCLC)
[121][105]. They also showed that the PARPis modulated the IFN-γ-induced PD-L1 expression in both NSCLC cell lines and patient tumor cells, an effect that was enhanced by ERCC1 deficiency. With cytotoxic doses of olaparib, Ding et al. showed that PARP inhibition induced strong local and systemic antitumor immune responses involving both CD4+ and CD8+ T cells in mice bearing BRCA1-deficient ovarian tumors
[122][106]. The authors demonstrated that the effect was driven by a STING-dependent type I IFN response from APCs that sensed either DNA fragments or cGAMP induced in the tumor cells. Using a genetically engineered mouse model of TNBC, Pantelidou et al. showed that Olaparib induced CD8+ T cell infiltration and activation that resulted in strong antitumor immunity
[123][107]. This response was diminished when CD8+ T cells were depleted, and was shown to be dependent on paracrine DC activation from cGAS-STING signaling in tumor cells. Using olaparib at doses in the IC50 range, the authors demonstrated that this effect was more prominent in HR-deficient than HR-proficient TNBC cells in vitro, and further confirmed this observation in vivo. Reisländer et al. demonstrated a dose-dependent induction of DNA DSB with olaparib in a H1229 NSCLC model possessing an inducible depletion of BRCA2
[124][108]. The BRCA2-deficient cells were more susceptible to olaparib induced DNA damage, which resulted in a rapid activation of an innate immune response involving increases in the mRNA levels of several interferon-stimulated genes (ISG). It is suggested that this response is conceivably from cGAS-STING activation; however, no direct evidence was provided. Shen et al. showed that cytostatic doses of talazoparib generated cytosolic dsDNA and activated cGAS-STING signaling in various gynecological cancer cell lines in vitro
[125][109]. This activity was further confirmed in vivo in a mouse ID8 ovarian cancer model where strong antitumor immunity was induced independent of BRCA status. The authors further showed that talazoparib treatment triggered ISG expression in orthotopic xenograft mouse models of BRCA1-deficient TNBC and BRCA2-deficient colorectal cancer.
2.2.3. Ataxia Telangiectasia and Rad3-Related (ATR) and ATM Inhibitors
ATR and ATM are critical signaling kinases in the DDR that are activated in response to DNA SSB and DSB, respectively
[126][127][137,138]. They have pivotal roles in coordinating the DDR and the cell cycle to prevent replications stress. ATR is recruited to replication protein A (RPA)-coated single-stranded DNA via the ATR-interacting protein (ATRIP)
[128][129][139,140], where it activates its major downstream effector, checkpoint kinase 1 (CHK1), which triggers intra-S and G2/M phase checkpoints
[130][141]. ATM is recruited to DSB by the MRN complex
[131][142], where it activates p53 by both phosphorylating it (Ser 15) and eliminating the inhibitory binding of the E3 ubiquitin ligase MDM2, which inhibits CDK2/cyclin E to induce cell cycle arrest at the G1 phase checkpoint
[126][137]. Many chemotherapeutic agents induce an activation of ATR, which has been the rationale for its therapeutic inhibition
[132][133][143,144].
While ATR and ATM inhibitors do not damage DNA directly, they have been shown to modulate immune responses induced by both radiotherapy
[134][135][136][137][110,111,145,146] and DNA-damaging chemotherapies
[138][139][147,148]. The combination treatment of radiotherapy and ATR or ATM inhibition was shown to induce both type I and type II IFN-associated gene responses, as well as increase CD8+ T cell infiltration
[134][135][136][137][110,111,145,146]. There is emerging evidence that this activity may be due, in part, to cGAS-STING activation. Sheng et al. showed that the ATR inhibitor ceralasertib (AZD6738) increased the degree of radiotherapy-induced CD8+ T cell infiltration and activation in a mouse xenograft model of hepatocellular carcinoma, while also decreasing the immunosuppressive effects of the radiation on numbers of intratumoral Tregs and exhausted T cells
[134][110]. The authors also showed that the addition of ATR inhibitor to an anti-PDL1 radioimmunotherapy program produced a cGAS-STING-dependent synergistic effect with increased infiltration, proliferation, and IFN-γ production from tumor infiltrating CD8+ T cells, as well as decreased numbers of tumor resident Tregs and exhausted T cells. Using the ATM inhibitor KU60019, Zhang et al. demonstrated a cooperative activity with radiotherapy to induce type I IFN signaling in human and mouse pancreatic cancer cell lines in a manner that was independent of cGAS-STING, but dependent on both TBK1 and the proto-oncogene tyrosine protein kinase SRC
[135][111]. The combination of ATM inhibition and radiotherapy was further shown to increase PDL-1 expression and tumor sensitivity to anti-PDL-1 therapy in a mouse model of pancreatic cancer, accompanied by an increased tumoral infiltration of CD8+ T cells that established immunological memory. Hu et al. showed a potent cGAS-STING pathway activation with the ATM inhibitors AZD1390 and KU55933 in both human breast cancer and mouse melanoma cells
[140][112]. The authors linked the resulting increase in phosphor-TBK1 levels and ISG expression with down-regulation of mitochondrial transcription factor A, and subsequent cytoplasmic leakage of mitochondrial DNA.