Alzheimer’s disease (AD) is the most common neurodegenerative disorder globally. In people aged 65 and older, it is estimated that 1 in 9 currently live with the disease. With aging being the greatest risk factor for disease onset, the physiological, social and economic burden continues to rise. Thus, AD remains a public health priority. Since 2007, genome-wide association studies (GWAS) have identified over 80 genomic loci with variants associated with increased AD risk. Although some variants are beginning to be characterized, the effects of many risk loci remain to be elucidated. One advancement which may help provide a patient-focused approach to tackle this issue is the application of gene editing technology and human-induced pluripotent stem cells (hiPSCs).
- Alzheimer’s disease
- GWAS
- iPSC
- microglia
- neurodegeneration
1. Introduction to Alzheimer’s Disease
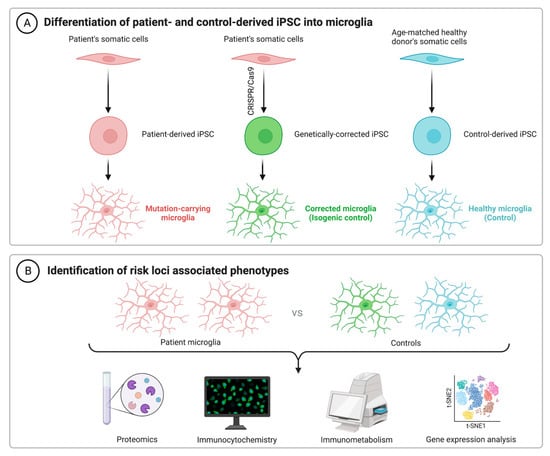
2. iPSC Technology and iPSC-Derived Microglia
As an alternative to using immortalized lines, primary cells from animal models and human microglia from brain excision surgery, recent advances in technology have facilitated a renewed interested in the generation of microglia from human-induced pluripotent stem cells (hiPSCs). Getting around the issue of tissue availability and variation in microglial phenotypes, hiPSCs can be used for both in vitro and xenographic in vivo work to understand cellular physiology in both health and disease. For example, microglial chimeric mouse models have been successfully generated, wherein hiPSC-derived macrophage progenitors are transplanted into neonatal mouse brains. Single-cell RNA sequencing reported that these xenografted cells largely retain human microglial identity and become widely dispersed by 6 months of age, including in both the hippocampus and cerebral cortex, as reported by TMEM119 staining [20][82]. It has been suggested that TMEM119 is both a reliable and specific marker, able to discriminate resident microglia from peripherally derived macrophages [21][22][83,84]. However, recent work suggests that TMEM119 is not exclusive, nor does it stain all microglia. The latter appears especially true under cellular stress conditions, including murine models of both focal stroke and Parkinson’s disease [23][85]. As such, supplementary staining methods will be important for future work using this model. Despite this issue though, in tandem with murine microglial depletion methods such as the pharmacological inhibition of CSF1R [24][25][86,87], the function of humanized microglia can be validated and compared against traditional non-chimeric animals. Pharmacological and genetic studies which report benefits in both systems will likely strengthen the translational potential of any promising future therapeutic target. Although the understanding of microglial function continues to improve, cellular dysfunction in AD remains to be fully elucidated, despite the identification of dozens of genetic risk loci (Table 1). The use of hiPSCs to decipher the pathological contributions of these risk variants could thus prove invaluable. Following the seminal publication by Muffet and colleagues in 2016 [26][88], many protocols to produce hiPSC-derived microglia have become available [27][28][29][30][31][32][89,90,91,92,93,94], with others having reviewed some of these protocols previously [33][95].| Gene | SNPs | GWAS Source |
|---|---|---|
| ABCA1 | rs1800978 | [35][63] |
| ABCA7 | rs12151021, rs3752231, rs3752246, rs4147929 | [35][36][37][38][39][63,64,69,70,71] |
| ABI3 | rs616338 | [35][63] |
| ACE | rs4277405, rs6504163 | [35][36][63,64] |
| ADAM10 | rs442495, rs593742 | [37][39][40][65,69,71] |
| ADAM17 | rs72777026 | [35][63] |
| ADAMTS1 | rs2830489, rs2830500 | [35][39][63,71] |
| ALPK2 | rs76726049 | [40][65] |
| ANKH | rs112403360 | [35][63] |
| APH1B | rs117618017 | [35][36][40][63,64,65] |
| APOE | rs429358 | [35][36][39][63,64,71] |
| BIN1 | rs4663105, rs6733839 | [35][36][37][38][39][40][63,64,65,69,70,71] |
| BLNK | rs6584063 | [35][63] |
| CASS4 | rs6014724, rs6024870, rs6069737, rs7274581 | [35][36][37][38][39][63,64,69,70,71] |
| CELF1 | rs10838725 | [38][70] |
| CD2AP | rs7767350, rs9369716, rs9381563, rs9473117, rs10948363 | [35][36][37[40][63,64][,6538][,6939],70,71] |
| CD33 | rs1354106, rs3865444, rs12459419 | [36][37][40][64,65,69] |
| CLNK | rs4504245, rs6448453, rs6846529 | [35][36][40][63,64,65] |
| CLU | rs1532278, rs4236673, rs9331896, rs11787077 | [35][36][37][38][39][40][63,64,65,69,70,71] |
| CNTNAP2 | rs114360492 | [40][65] |
| COX7C | rs62374257 | [35][63] |
| CR1 | rs679515, rs2093760, rs4844610, rs6656401 | [35][36][37][38][39][40][63,64,65,69,70,71] |
| CSTF1 | rs6069736 | [37][69] |
| CTSB | rs1065712 | [35][63] |
| CTSH | rs12592898 | [35][63] |
| CYB561 | rs138190086 | [37][39][69,71] |
| DOC2A | rs1140239 | [35][63] |
| ECHDC3 | rs7920721 | [37][39][69,71] |
| EED | rs3851179 | [35][39][63,71] |
| EPHA1 | rs3935067, rs7810606, rs10808026, rs11771145 | [35][36][37][38][39][40][63,64,65,69,70,71] |
| FERMT2 | rs7146179, rs17125924, rs17125944 | [35][36][37][38][39][63,64,69,70,71] |
| FOXF1 | rs16941239 | [35][63] |
| GPR141 | rs2718058 | [38][70] |
| GRN | rs5848 | [35][63] |
| HESX1 | rs184384746 | [40][65] |
| HLA-DQA1 | rs1846190, rs6605556, rs6931277, rs9271192 | [35][36][38][40][63,64,65,70] |
| HLA-DRB1 | rs9271058 | [39][71] |
| ICA1 | rs10952097 | [35][63] |
| IL34 | rs4985556 | [35][63[37],69] |
| INPP5D | rs7597763, rs10933431, rs35349669 | [35][36][37][38][39][40][63,64,65,69,70,71] |
| IQCK | rs7185636 | [39][71] |
| MEF2C | rs190982 | [38][70] |
| MINDY2 | rs602602 | [35][36][63,64] |
| MME | rs16824536, rs61762319 | [35][63] |
| MS4A4A | rs1582763, rs2081545 | [35][36][37][40][63,64,65,69] |
| MS4A6A | rs983392, rs7933202 | [38][39][70,71] |
| MYO15A | rs2242595 | [35][63] |
| NCK2 | rs115186657, rs143080277 | [36][40][64,65] |
| NECTIN2 | rs41289512 | [37][40][65,69] |
| NYAP1 | rs12539172 | [39][71] |
| OARD1 | rs114812713 | [39][71] |
| PICALM | rs867611, rs561655, rs10792832 | [36][37][38][64[,6540,69],70] |
| PLCG2 | rs12444183, rs12446759, rs72824905 | [35][37][63,69] |
| PRDM7 | rs56407236 | [35][63] |
| PRKD3 | rs17020490 | [35][63] |
| PSMC3 | rs12292911 | [37][69] |
| PTK2B | rs28834970, rs73223431 | [35][38][39][63,70,71] |
| RASGEF1C | rs113706587 | [35][63] |
| SCIMP | rs7225151, rs113260531 | [35][37][40][63,65,69] |
| SEC61G | rs76928645 | [35][63] |
| SHARPIN | rs34173062 | [35][63] |
| SLC24A4 | rs7401792, rs10498633, rs12590654, rs12881735 | [35][36][37][38][39][40][63,64,65,69,70,71] |
| SORL1 | rs11218343, rs74685827 | [35][36][37][38][39][40][63,64,65,69,70,71] |
| SORT1 | rs141749679 | [35][63] |
| SPDYE3 | rs7384878 | [35][36][63,64] |
| SPI1 | rs3740688, rs10437655 | [35][36][39][63,64,71] |
| SPPL2A | rs8025980, rs59685680 | [35][37][63,69] |
| TMEM121 | rs7157106, rs10131280 | [35][63] |
| TPCN1 | rs6489896 | [35][63] |
| TREM2 | rs75932628, rs143332484 | [35][39][63,71] |
| TREML2 | rs9381040, rs60755019 | [35][37][63,69] |
| TSPAN14 | rs6586028 | [35][63] |
| UMAD1 | rs6943429 | [35][63] |
| UNC5CL | rs10947943, rs187370608 | [35][36][40][63,64,65] |
| USP6NL | rs7912495, rs11257238 | [35][36][40][63,64,65] |
| WDR12 | rs139643391 | [35][63] |
| WDR81 | rs35048651 | [35][63] |
| WNT3 | rs199515 | [35][63] |
| WWOX | rs62039712 | [39][71] |
| ZCWPW1 | rs1476679 | [37][38][69,70] |
| ZNF652 | rs28394864 | [36][40][64,65] |