Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Rita Xu and Version 1 by Chin-Kun Wang.
Phytochemicals are plant secondary metabolites that show health benefits for humans due to their bioactivity. There is a huge variety of phytochemicals that have already been identified, and these compounds can act as antimicrobial and neuroprotection agents. Due to their anti-microbial activity and neuroprotection, several phytochemicals might have the potency to be used as natural therapeutic agents, especially for
Helicobacter pylori
infection and neurodegenerative disease, which have become a global health concern nowadays.
- phytochemical
- neurodegenerative disease
- Helicobacter pylori
1. Introduction
Helicobacter pylori infection is one of the global health problems. More than 50% of the population in the world is affected, mostly in developing countries [1]. H. pylori attaches to the human stomach; induces a change in gastric physiology; and is highly associated with gastric ulcers, which further progress into gastric cancer [2]. H. pylori can colonize and infect gastric tissue because of virulent factors such as urease, lipopolysaccharide (LPS), vacuolating cytotoxin A (VacA), cytotoxin-associated gene A (CagA), and some others [3]. Until now, the main treatment for H. pylori infection is to use the combination of two antibiotics together with a bismuth compound and/or antacid agent such proton pump inhibitor (PPI), which is called quadruple therapy and provides an eradication rate of more than 80% [4]. The usage of antibiotics in H. pylori offers another concern of some side effects as well as antibiotic resistance problems [5]. Recent studies show that H. pylori infection contributes to the progression of neurodegenerative diseases.
Neurodegenerative diseases (NDs) are disorders that affect the central nervous system and that are mostly caused by neuronal cell death, which causes impairment of the cognitive and motoric system [6]. There are many risk factors associated with ND progression, but its pathogenesis has still been unclear until now. Several diseases are classified as NDs such as Alzheimer’s disease (AD), Parkinson’s disease (PD), and Huntington’s disease (HD) [7]. These diseases have different characteristics, but most of them share the same hallmarks, which are neuronal cell death and neuroinflammation [8,9][8][9]. Until now, ND has been classified as an incurable disease, and medication might have a small impact on improving a patient’s condition [10]. Evidence of nutraceuticals on NDs is still deficient, in terms of whether together with normal medication, they could provide better effects on subjects with NDs.
There are several hypotheses about the possible connection between H. pylori infection and NDs. H. pylori affect the absorption of folate and vitamin B-12, which causes the elevation of homocysteine level and induces neurotoxicity. Furthermore, H. pylori cross the blood–brain barrier and induce amyloid deposition in the brain [11]. Another study showed that the outer membrane vesicles of H. pylori that were injected into mice altered astrocyte function and induced neuronal damage in the mouse brain [12]. In PD, it showed that H. pylori infection is related to the progression of the disease and increases the requirement of medication for PD [13]. This evidence might provide a clue about the connection between neurodegenerative disease and H. pylori infection.
Phytochemicals are secondary metabolites of plants, which are non-nutritive bio-active compounds synthesized for natural defenses of the plant against pests [14,15,16,17][14][15][16][17]. Phytochemicals found in fruits, vegetables, nuts, and grains provide health benefits. Many studies showed that phytochemicals from different natural sources act as antibacterial agents or neuroprotective agents [17,18][17][18]. Önem et al. showed that stalk extracts from two different cultivars of Prunus avium L. inhibited Gram-positive bacteria and reduced the biofilm formation of bacteria by up to 75% [19]. Li et al. showed that supplementation of proanthocyanidins (PAC)-rich cranberry juice (44 mg of PAC per portion) twice a day for 8 weeks significantly reduced H. pylori infection [20]. Desideri et al. reported that high intake (990 mg/day) of dietary flavonols from cocoa for 8 weeks significantly improved cognitive function in mild cognitive impairment subjects compared to those of low intake (45 mg/day) of cocoa flavonols [21]. Kent et al. pointed out that the intervention of anthocyanin-rich cherry juice for 12 weeks significantly improved verbal fluency and short-term and long-term memory in subjects with dementia [22]. Past studies showed that phytochemicals can be used as drug alternatives to treat H. pylori and neurodegenerative disease and reduce the risk of antibiotic resistance and complications due to the medication.
2. H. pylori
H. pylori is a Gram-negative spiral bacterium that is found in the human stomach and is associated with gastric ulcer and advanced gastric cancer [2,23,24][2][23][24]. The infection of H. pylori shows no symptoms in most cases, but it depends on the immune response of the individual and the severity of the syndrome. Most symptoms of H. pylori infection are correlated with the gastric ulcer and inflammation in the gastric tissue [25]. H. pylori is considered a special bacterium due to the virulence factors (Figure 1) that help to colonize in the human stomach, such as VacA, CagA, urease, LPS, and different kinds of adhesins [3].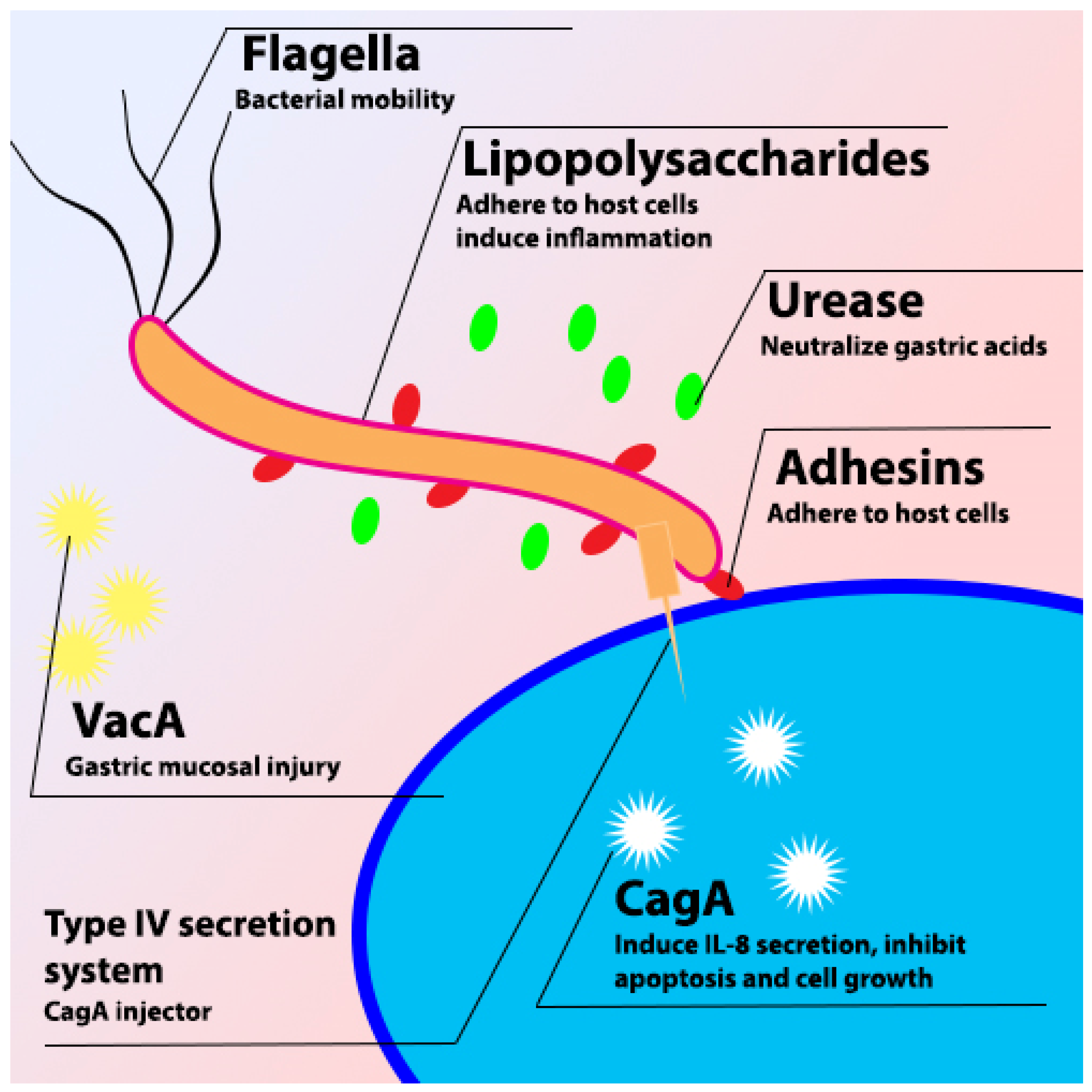
Figure 1. Schematic diagram of H. pylori virulence factor.
2.1. VacA
VacA is one of the virulence factors possessed by H. pylori. VacA is the major toxic 88 kDa protein that is secreted from H. pylori through type V auto transport secretion system (T5SS), which binds to the host cell and causes vacuolation of the cell [26]. VacA plays an important role in the colonization of H. pylori in the gastric mucosa, stimulating the autophagy pathway in cells and disrupting lysosomal trafficking that causes the accumulation of dysfunctional autophagosomes and the formation of large intracellular vacuoles to promote the intracellular survival of H. pylori [27]. Furthermore, it induces different responses in infected cells such as vacuole formation, cytochrome c release, and forming channels in the mitochondria [28]. It also induces cell apoptosis because of increasing cytochrome c release from mitochondria. Cytochrome c combines with Apaf-1 and caspase-9 to stimulate the production of caspase-3 and caspase-7, resulting in cell apoptosis [29,30][29][30]. VacA can disrupt the tight junction to alter the tissue structure and increase the adhesion of H. pylori to epithelial cells [26,31,32][26][31][32].2.2. CagA
CagA is a 120 to 145 kDa protein that can be injected into the host cell by using a type IV secretion system (T4SS) after the adhesion of H. pylori to the host cell [33]. H. pylori is divided into two different strains based on the presence of CagA: CagA-positive and CagA-negative strains. The cagA-positive strain is more virulent than the CagA-negative strain and is associated with higher gastric inflammation [34]. The effects of CagA on the host cell are independent of the phosphorylation process. The most noticeable is to disrupt the cell’s tight junction and induce cell morphology changes [35]. Non-phosphorylated CagA also can activate serum response elements further affect the cell cycle and induce inflammatory response [36].2.3. Urease
Urease is a 550 kDa molecule consisting of UreA and UreB subunits. Urease plays a crucial role in the survival of H. pylori in the human stomach. H. pylori produces urease in acidic conditions, which breaks down urea and releases ammonia to neutralize the acidic condition in the human stomach [37]. pH increases in the stomach alter the protective mucous layer and also dysregulate the gastric epithelial cell tight junction [38].2.4. Pathophysiology of H. pylori Infection
H. pylori infection is associated with chronic gastritis, gastric ulcers, and gastric cancer [1]. Development of gastric problems due to H. pylori infection is mostly caused by alteration of the gastric physiology and microenvironment, which induces an immune response from the human body [39]. This immune response is due to the activity of the H. pylori virulence factors such as CagA, VacA, and urease, and the response might be different depending on the age [40,41][40][41]. Immune response due to H. pylori infection is mediated by Toll-like receptors (TLRs) and microRNA, which can promote or suppress the immune response [42]. After reaching the stomach, H. pylori move to the mucous layer to evade the acid condition with the help of urease and attach to epithelial cells with the help of different kinds of adhesins such as BabA, SabA, AlpA/B, HopZ, and OipA [43]. After binding to the host cell, H. pylori inject different kinds of toxins such as CagA and VacA, depending on the strain, being able to induce inflammatory responses and upregulation of pro-inflammatory cytokines secretion [1].2.5. Diagnosis and Treatment
There are various methods to identify and diagnose H. pylori. The invasive tests are based on gastric biopsy and peripheral samples to check the infection of H. pylori. On the other hand, the non-invasive method is to use the Urea Breath Test (UBT) C13 or C14 [1]. UBT is one of the most popular methods to diagnose H. pylori infection due to its high sensitivity and is considered the gold standard of the non-invasive method [1]. UBT is based on the reaction of C13-labeled urea and bacterial urease secreted from H. pylori, which produce ammonia (NH3) and C13-labeled carbon dioxide in the breath. The concentration of the C13 isotope is determined by using gas chromatography and considered positive if the Delta Over Baseline (DOB) value is ≥4‰ [44,45,46][44][45][46]. Treatment of H. pylori infection is usually conducted by using antibiotics and combination with PPI and/or with bismuth. Monotherapy (single antibiotic) was used in the past, but the efficacy was poor. The addition of PPI is used as dual therapy in some countries. Overuse of antibiotics induces the mutation and resistance of H. pylori and produces some side effects such as dizziness, vomiting, and allergy [47,48][47][48].3. Connection between H. pylori Infection and Neurodegenerative Diseases
There are several risk factors correlated with NDs, especially AD, such as age, traumatic head injury, depression, cardiovascular and cerebrovascular disease, and smoking. Recent studies showed that AD is also associated with H. pylori infection [91,92][49][50]. H. pylori is known to infect and cause several health problems in the human gastrointestinal (GI) tract such as gastric ulcers, gastritis, and gastric cancer [93][51]. In rare cases, a manifestation of extra gastric disease due to H. pylori infection might occur with several possible mechanism (Figure 2) and need to be taken into consideration. The extra gastric manifestation due to H. pylori infection, especially neurological problems, might occur through alteration of the gut–brain axis (GBA) [94][52]. The GBA is a bidirectional communication between the central nervous system (CNS) and enteric nervous system that integrates and links the gut and intestinal function with the central nervous system [95,96,97][53][54][55]. GBA modulates the GI function by regulating the GI immune system, mucosal change, and intestinal microbiome in response to stress and emotional and environmental influences [94,98][52][56].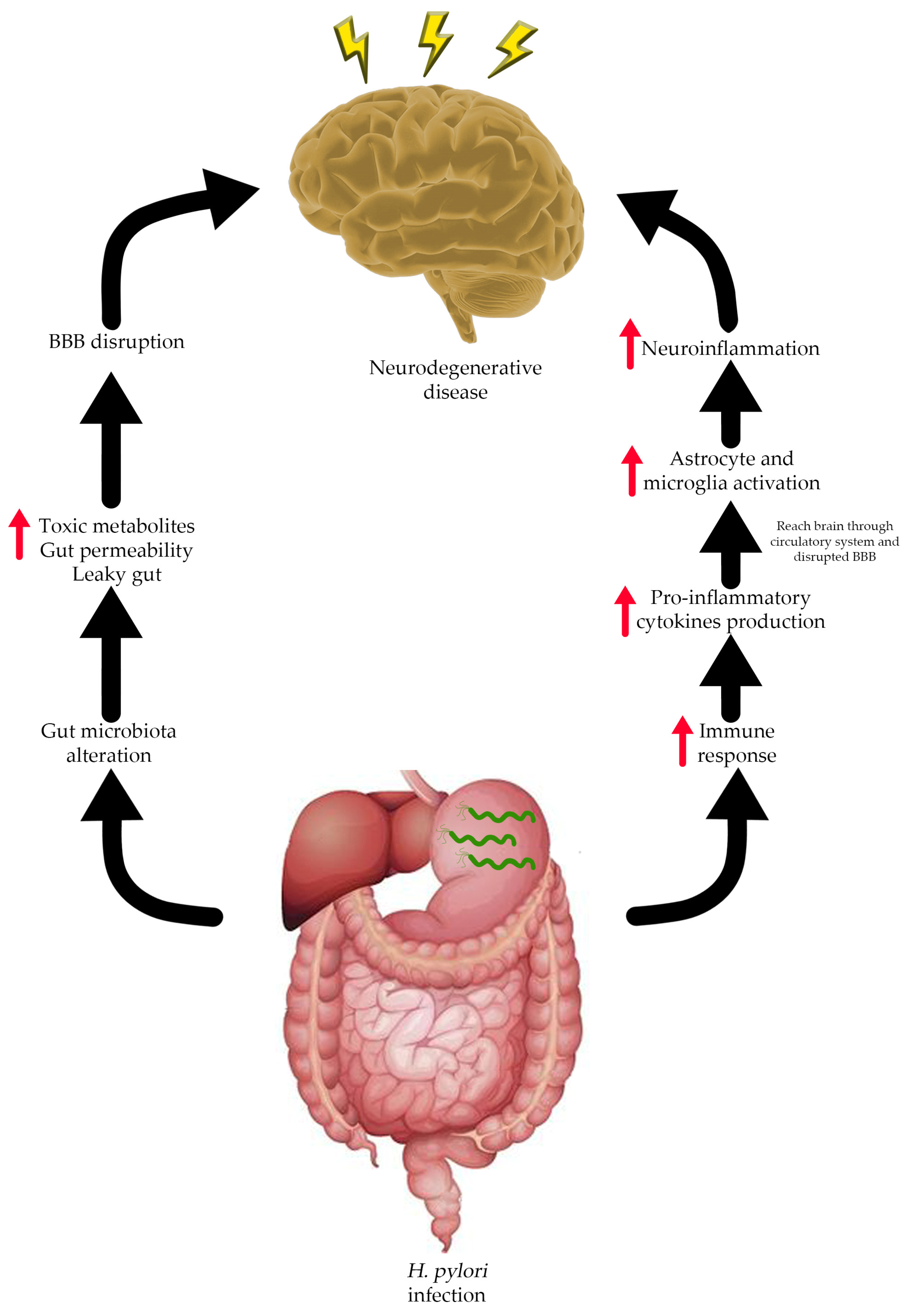
Figure 2. Possible relationship between H. pylori infection and neurodegenerative disease.
References
- Kusters, J.G.; van Vliet, A.H.M.; Kuipers, E.J. Pathogenesis of Helicobacter pylori infection. Clin. Microbiol. Rev. 2006, 19, 449–490.
- Dunn, B.E.; Cohen, H.; Blaser, M.J. Helicobacter pylori . Clin. Microbiol. Rev. 1997, 10, 720–741.
- Kao, C.-Y.; Sheu, B.-S.; Wu, J.-J. Helicobacter pylori infection: An overview of bacterial virulence factors and pathogenesis. Biomed. J. 2016, 39, 14–23.
- Selgrad, M.; Malfertheiner, P. Treatment of Helicobacter pylori. Curr. Opin. Gastroenterol. 2011, 27, 565–570.
- Bytzer, P.; Dahlerup, J.F.; Eriksen, J.R.; Jarbøl, D.E.; Rosenstock, S.; Wildt, S.; Danish Society for Gastroenterology. Diagnosis and treatment of Helicobacter pylori infection. Dan. Med. Bull. 2011, 58, C4271.
- Dugger, B.N.; Dickson, D.W. Pathology of neurodegenerative diseases. Cold Spring Harb. Perspect. Biol. 2017, 9, a028035.
- Gitler, A.D.; Dhillon, P.; Shorter, J. Neurodegenerative disease: Models, mechanisms, and a new hope. Dis. Model Mech. 2017, 10, 499–502.
- Chi, H.; Chang, H.-Y.; Sang, T.-K. Neuronal cell death mechanisms in major neurodegenerative diseases. Int. J. Mol. Sci. 2018, 19, 3082.
- Zhang, W.; Xiao, D.; Mao, Q.; Xia, H. Role of neuroinflammation in neurodegeneration development. Signal Transduct. Target Ther. 2023, 8, 267.
- Mariani, E.; Polidori, M.C.; Cherubini, A.; Mecocci, P. Oxidative stress in brain aging, neurodegenerative and vascular diseases: An overview. J. Chromatogr. B. Analyt. Technol. Biomed. Life Sci. 2005, 827, 65–75.
- Beydoun, M.A.; Beydoun, H.A.; Elbejjani, M.; Dore, G.A.; Zonderman, A.B. Helicobacter pylori seropositivity and its association with incident all-cause and Alzheimer’s disease dementia in large national surveys. Alzheimer’s Dement. 2018, 14, 1148–1158.
- Palacios, E.; Lobos-González, L.; Guerrero, S.; Kogan, M.J.; Shao, B.; Heinecke, J.W.; Quest, A.F.G.; Leyton, L.; Valenzuela-Valderrama, M. Helicobacter pylori outer membrane vesicles induce astrocyte reactivity through nuclear factor-κappa b activation and cause neuronal damage in vivo in a murine model. J. Neuroinflamm. 2023, 20, 66.
- Mridula, K.R.; Borgohain, R.; Chandrasekhar Reddy, V.; Srinivasarao Bandaru, V.C.; Suryaprabha, T. Association of Helicobacter pylori with Parkinson’s disease. J. Clin. Neurol. 2017, 13, 181.
- AL-Ishaq, R.K.; Overy, A.J.; Büsselberg, D. Phytochemicals and gastrointestinal cancer: Cellular mechanisms and effects to change cancer progression. Biomolecules 2020, 10, 105.
- Shu, L.; Cheung, K.-L.; Khor, T.O.; Chen, C.; Kong, A.-N. Phytochemicals: Cancer chemoprevention and suppression of tumor onset and metastasis. Cancer Metastasis Rev. 2010, 29, 483–502.
- Petrovska, B.B. Historical review of medicinal plants′ usage. Pharmacogn. Rev. 2012, 6, 1.
- Lima, M.C.; Paiva de Sousa, C.; Fernandez-Prada, C.; Harel, J.; Dubreuil, J.D.; de Souza, E.L. A review of the current evidence of fruit phenolic compounds as potential antimicrobials against pathogenic bacteria. Microb. Pathog. 2019, 130, 259–270.
- Gregory, J.; Vengalasetti, Y.V.; Bredesen, D.E.; Rao, R.V. Neuroprotective herbs for the management of Alzheimer’s disease. Biomolecules 2021, 11, 543.
- Önem, E.; Sarısu, H.C.; Özaydın, A.G.; Muhammed, M.T.; Ak, A. Phytochemical profile, antimicrobial, and anti-quorum sensing properties of fruit stalks of Prunus Avium L. Lett. Appl. Microbiol. 2021, 73, 426–437.
- Li, Z.; Ma, J.; Guo, Y.; Liu, W.; Li, M.; Zhang, L.; Zhang, Y.; Zhou, T.; Zhang, J.; Gao, H.; et al. Suppression of Helicobacter pylori infection by daily cranberry intake: A double-blind, randomized, placebo-controlled trial. J. Gastroenterol. Hepatol. 2020, 36, 927–935.
- Desideri, G.; Kwik-Uribe, C.; Grassi, D.; Necozione, S.; Ghiadoni, L.; Mastroiacovo, D.; Raffaele, A.; Ferri, L.; Bocale, R.; Lechiara, M.; et al. Benefits in cognitive function, blood pressure, and insulin resistance through cocoa flavanol consumption in elderly subjects with mild cognitive impairment: The cocoa, cognition, and aging (cocoa) study. Hypertension 2012, 60, 794–801.
- Kent, K.; Charlton, K.; Roodenrys, S.; Batterham, M.; Potter, J.; Traynor, V.; Gilbert, H.; Morgan, O.; Richards, R. Consumption of anthocyanin-rich cherry juice for 12 weeks improves memory and cognition in older adults with mild-to-moderate dementia. Eur. J. Nutr. 2015, 56, 333–341.
- Marshall, B.J.; Armstrong, J.A.; McGechie, D.B.; Clancy, R.J. Attempt to fulfil Koch’s postulates for pyloric Campylobacter. Med. J. Aust. 1985, 142, 436–439.
- Gotteland, M.; Andrews, M.; Toledo, M.; Muñoz, L.; Caceres, P.; Anziani, A.; Wittig, E.; Speisky, H.; Salazar, G. Modulation of Helicobacter pylori colonization with cranberry juice and Lactobacillus johnsonii La1 in children. Nutrition 2008, 24, 421–426.
- Abbas, M.; Sharif, F.A.; Osman, S.M.; Osman, A.M.; El Sanousi, S.M.; Magzoub, M.; Ibrahim, M.E. Prevalence and associated symptoms of Helicobacter pylori infection among schoolchildren in Kassala state, east of Sudan. Interdiscip. Perspect. Infect. Dis. 2018, 2018, 4325752.
- Palframan, S.L.; Kwok, T.; Gabriel, K. Vacuolating cytotoxin a (VacA), a key toxin for Helicobacter pylori pathogenesis. Front. Cell. Infect. Microbiol. 2012, 2, 92.
- Abdullah, M.; Greenfield, L.K.; Bronte-Tinkew, D.; Capurro, M.I.; Rizzuti, D.; Jones, N.L. VacA promotes CagA accumulation in gastric epithelial cells during Helicobacter pylori Infection. Sci. Rep. 2019, 9, 38.
- Willhite, D.C.; Cover, T.L.; Blanke, S.R. Cellular vacuolation and mitochondrial cytochrome c release are independent outcomes of Helicobacter pylori vacuolating cytotoxin activity that are each dependent on membrane channel formation. J. Biol. Chem. 2003, 278, 48204–48209.
- Rao, R.V.; Peel, A.; Logvinova, A.; del Rio, G.; Hermel, E.; Yokota, T.; Goldsmith, P.C.; Ellerby, L.M.; Ellerby, H.M.; Bredesen, D.E. Coupling endoplasmic reticulum stress to the cell death program: Role of the er chaperone grp78. FEBS Lett. 2002, 514, 122–128.
- Fulda, S.; Debatin, K.-M. Extrinsic versus intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene 2006, 25, 4798–4811.
- Rassow, J. Helicobacter pylori vacuolating toxin A and apoptosis. Cell Commun. Signal. 2011, 9, 26.
- Foegeding, N.J.; Caston, R.R.; McClain, M.S.; Ohi, M.D.; Cover, T.L. An overview of Helicobacter pylori VacA toxin biology. Toxins 2016, 8, 173.
- Jiménez-Soto, L.F.; Haas, R. The CagA toxin of Helicobacter pylori: Abundant production but relatively low amount translocated. Sci. Rep. 2016, 6, 23227.
- Hatakeyama, M. Structure and function of Helicobacter pylori CagA, the first-identified bacterial protein involved in human cancer. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2017, 93, 196–219.
- Jones, K.R.; Whitmire, J.M.; Merrell, D.S. A Tale of two toxins: Helicobacter pylori CagA and VacA modulate host pathways that impact disease. Front. Microbiol. 2010, 1, 23227.
- Suzuki, M.; Mimuro, H.; Kiga, K.; Fukumatsu, M.; Ishijima, N.; Morikawa, H.; Nagai, S.; Koyasu, S.; Gilman, R.H.; Kersulyte, D.; et al. Helicobacter pylori CagA phosphorylation-independent function in epithelial proliferation and inflammation. Cell Host Microbe 2009, 5, 23–34.
- Mobley, H.; The role of Helicobacter pylori urease in the pathogenesis of gastritis and peptic ulceration. Aliment. Pharmacol. Ther. 1996, 10 (Suppl. S1), 57–64.
- Olivera-Severo, D.; Uberti, A.F.; Marques, M.S.; Pinto, M.T.; Gomez-Lazaro, M.; Figueiredo, C.; Leite, M.; Carlini, C.R. A New Role for Helicobacter pylori urease: Contributions to angiogenesis. Front. Microbiol. 2017, 8, 1883.
- Xu, W.; Xu, L.; Xu, C. Relationship between Helicobacter pylori infection and gastrointestinal microecology. Front. Cell. Infect. Microbiol. 2022, 12, 938608.
- Mišak, Z.; Hojsak, I.; Homan, M. Review: Helicobacter pylori in pediatrics. Helicobacter 2019, 24, e12639.
- Araújo, G.R.L.; Marques, H.S.; Santos, M.L.C.; da Silva, F.A.F.; de Brito, B.B.; Santos, G.L.C.; de Melo, F.F. Helicobacter pylori infection: How does age influence the inflammatory pattern? World J. Gastroenterol. 2022, 28, 402–411.
- Meliț, L.E.; Mărginean, C.O.; Mărginean, C.D.; Mărginean, M.O. The relationship between toll-like receptors and Helicobacter pylori-related gastropathies: Still a controversial topic. J. Immunol. Res. 2019, 2019, 8197048.
- Kalali, B.; Mejías-Luque, R.; Javaheri, A.; Gerhard, M. H. pylori Virulence factors: Influence on immune system and pathology. Mediators Inflamm. 2014, 2014, 426309.
- Logan, R.P. Urea Breath Tests in the Management of Helicobacter pylori infection. Gut 1998, 43 (Suppl. S1), S47–S50.
- Yang, H.; Hu, B. Diagnosis of Helicobacter pylori infection and recent advances. Diagnostics 2021, 11, 1305.
- Graham, D.Y.; Miftahussurur, M. Helicobacter pylori urease for diagnosis of Helicobacter pylori infection: A mini review. J. Adv. Res. 2018, 13, 51–57.
- Blumenthal, K.G.; Peter, J.G.; Trubiano, J.A.; Phillips, E.J. Antibiotic allergy. Lancet 2019, 393, 183–198.
- Patangia, D.V.; Anthony Ryan, C.; Dempsey, E.; Paul Ross, R.; Stanton, C. Impact of antibiotics on the human microbiome and consequences for host health. Microbiologyopen 2022, 11, e1260.
- Doulberis, M.; Saleh, C.; Beyenburg, S. Is There an association between migraine and gastrointestinal disorders? J. Clin. Neurol. 2017, 13, 215.
- Kountouras, J.M.D.P.; Tsolaki, M.; Gavalas, E.; Boziki, M.; Zavos, C.; Karatzoglou, P.; Chatzopoulos, D.; Venizelos, I. Relationship between Helicobacter pylori infection and Alzheimer disease. Neurology 2006, 66, 938–940.
- Hooi, J.K.Y.; Lai, W.Y.; Ng, W.K.; Suen, M.M.Y.; Underwood, F.E.; Tanyingoh, D.; Malfertheiner, P.; Graham, D.Y.; Wong, V.W.S.; Wu, J.C.Y.; et al. Global prevalence of Helicobacter pylori infection: Systematic Review and Meta-Analysis. Gastroenterology 2017, 153, 420–429.
- Budzyński, J.; Kłopocka, M. Brain-gut axis in the pathogenesis of Helicobacter pylori infection. World J. Gastroenterol. 2014, 20, 5212.
- Carabotti, M.; Scirocco, A.; Maselli, M.A.; Severi, C. The gut-brain axis: Interactions between enteric microbiota, central and enteric nervous systems. Ann. Gastroenterol. 2015, 28, 203–209.
- Zhou, L.; Foster, J.A. Psychobiotics and the gut–brain axis: In the pursuit of happiness. Neuropsychiatr. Dis. Treat. 2015, 11, 715.
- Morais, L.H.; Schreiber, H.L.; Mazmanian, S.K. The gut microbiota–brain axis in behaviour and brain disorders. Nat. Rev. Microbiol. 2020, 19, 241–255.
- Megur, A.; Baltriukienė, D.; Bukelskienė, V.; Burokas, A. The microbiota–gut–brain axis and Alzheimer’s disease: Neuroinflammation is to blame? Nutrients 2020, 13, 37.
- Iino, C.; Shimoyama, T. Impact of Helicobacter pylori infection on gut microbiota. World J. Gastroenterol. 2021, 27, 6224–6230.
- Yang, L.; Zhang, J.; Xu, J.; Wei, X.; Yang, J.; Liu, Y.; Li, H.; Zhao, C.; Wang, Y.; Zhang, L.; et al. Helicobacter pylori infection aggravates dysbiosis of gut microbiome in children with gastritis. Front. Cell. Infect. Microbiol. 2019, 9, 375.
- Zheng, W.; Miao, J.; Luo, L.; Long, G.; Chen, B.; Shu, X.; Gu, W.; Peng, K.; Li, F.; Zhao, H.; et al. The effects of Helicobacter pylori infection on microbiota associated with gastric mucosa and immune factors in children. Front. Immunol. 2021, 12, 625586.
- Shen, L.; Liu, L.; Ji, H.-F. Alzheimer’s disease histological and behavioral manifestations in transgenic mice correlate with specific gut microbiome state. J. Alzheimer’s Dis. 2017, 56, 385–390.
- Zou, B.; Li, J.; Ma, R.-X.; Cheng, X.-Y.; Ma, R.-Y.; Zhou, T.-Y.; Wu, Z.-Q.; Yao, Y.; Li, J. Gut microbiota is an impact factor based on the brain-gut axis to alzheimer’s disease: A systematic review. Aging Dis. 2023, 14, 964.
- Doulberis, M.; Kotronis, G.; Thomann, R.; Polyzos, S.A.; Boziki, M.; Gialamprinou, D.; Deretzi, G.; Katsinelos, P.; Kountouras, J. Review: Impact of Helicobacter pylori on Alzheimer’s disease: What do we know so far? Helicobacter 2017, 23, e12454.
- Tawfik, A.; Samra, Y.A.; Elsherbiny, N.M.; Al-Shabrawey, M. Implication of hyperhomocysteinemia in blood retinal barrier (BRB) dysfunction. Biomolecules 2020, 10, E1119.
- Brustolin, S.; Giugliani, R.; Félix, T.M. Genetics of homocysteine metabolism and associated disorders. Braz. J. Med. Biol. Res. 2010, 43, 1–7.
- Kumar, A.; Palfrey, H.A.; Pathak, R.; Kadowitz, P.J.; Gettys, T.W.; Murthy, S.N. The metabolism and significance of homocysteine in nutrition and health. Nutr. Metab. 2017, 14, 78.
- Selhub, J. Homocysteine metabolism. Annu. Rev. Nutr. 1999, 19, 217–246.
- Tinelli, C.; Di Pino, A.; Ficulle, E.; Marcelli, S.; Feligioni, M. Hyperhomocysteinemia as a risk factor and potential nutraceutical target for certain pathologies. Front. Nutr. 2019, 6, 49.
- Smith, A.D.; Refsum, H.; Bottiglieri, T.; Fenech, M.; Hooshmand, B.; McCaddon, A.; Miller, J.W.; Rosenberg, I.H.; Obeid, R. Homocysteine and dementia: An international consensus statement. J. Alzheimer’s Dis. 2018, 62, 561–570.
- Kountouras, J.; Gavalas, E.; Boziki, M.; Zavos, C. Helicobacter pylori may be involved in cognitive impairment and dementia development through induction of atrophic gastritis, vitamin b-12–folate deficiency, and hyperhomocysteinemia sequence. Am. J. Clin. Nutr. 2007, 86, 805–806.
- Kountouras, J.; Gavalas, E.; Zavos, C.; Stergiopoulos, C.; Chatzopoulos, D.; Kapetanakis, N.; Gisakis, D. Alzheimer’s disease and Helicobacter pylori infection: Defective immune regulation and apoptosis as proposed common links. Med. Hypotheses 2007, 68, 378–388.
- Albaret, G.; Sifré, E.; Floch, P.; Laye, S.; Aubert, A.; Dubus, P.; Azzi-Martin, L.; Giese, A.; Salles, N.; Mégraud, F.; et al. Alzheimer’s disease and Helicobacter pylori infection: Inflammation from stomach to brain? J. Alzheimer’s Dis. 2020, 73, 801–809.
- Malaguarnera, M.; Bella, R.; Alagona, G.; Ferri, R.; Carnemolla, A.; Pennisi, G. Helicobacter pylori and Alzheimer’s disease: A possible link. Eur. J. Intern. Med. 2004, 15, 381–386.
- Roubaud-Baudron, C.; Krolak-Salmon, P.; Quadrio, I.; Mégraud, F.; Salles, N. Impact of chronic Helicobacter pylori infection on Alzheimer’s disease: Preliminary results. Neurobiol. Aging 2012, 33, 1009.e11–1009.e19.
- Wang, X.-L.; Zeng, J.; Yang, Y.; Xiong, Y.; Zhang, Z.-H.; Qiu, M.; Yan, X.; Sun, X.-Y.; Tuo, Q.-Z.; Liu, R.; et al. Helicobacter pylori filtrate induces Alzheimer-like tau hyperphosphorylation by activating glycogen synthase kinase-3β. J. Alzheimer’s Dis. 2014, 43, 153–165.
- Rizzatti, G.; Lopetuso, L.R.; Gibiino, G.; Binda, C.; Gasbarrini, A. Proteobacteria: A common factor in human diseases. Biomed Res. Int. 2017, 2017, 9351507.
- Toledo, A.R.L.; Monroy, G.R.; Salazar, F.E.; Lee, J.-Y.; Jain, S.; Yadav, H.; Borlongan, C.V. Gut–brain axis as a pathological and therapeutic target for neurodegenerative disorders. Int. J. Mol. Sci. 2022, 23, 1184.
- Rhee, S.H. Lipopolysaccharide: Basic biochemistry, intracellular signaling, and physiological impacts in the gut. Intest. Res. 2014, 12, 90–95.
- Zielen, S.; Trischler, J.; Schubert, R. Lipopolysaccharide challenge: Immunological effects and safety in humans. Expert Rev. Clin. Immunol. 2015, 11, 409–418.
- Lubomski, M.; Tan, A.H.; Lim, S.-Y.; Holmes, A.J.; Davis, R.L.; Sue, C.M. Parkinson’s disease and the gastrointestinal microbiome. J. Neurol. 2019, 267, 2507–2523.
- Çamcı, G.; Oğuz, S. Association between Parkinson’s disease and Helicobacter pylori. J. Clin. Neurol. 2016, 12, 147.
- Lahner, E.; Annibale, B.; Delle Fave, G. Systematic review: Heliocobacter pylori infection and impaired drug absorption. Aliment. Pharmacol. Ther. 2009, 29, 379–386.
More