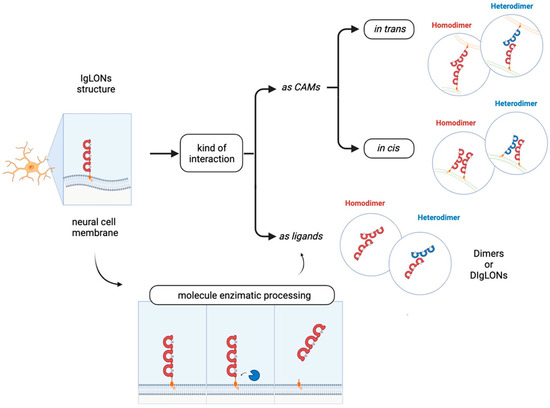
In the brain, cell adhesion molecules (CAMs) are critical for neurite outgrowth, axonal fasciculation, neuronal survival and migration, and synapse formation and maintenance. Among CAMs, the IgLON family comprises five members: Opioid Binding Protein/Cell Adhesion Molecule Like (OPCML or OBCAM), Limbic System Associated Membrane Protein (LSAMP), neurotrimin (NTM), Neuronal Growth Regulator 1 (NEGR1), and IgLON5. IgLONs exhibit three N-terminal C2 immunoglobulin domains; several glycosylation sites; and a glycosylphosphatidylinositol anchoring to the membrane. Interactions as homo- or heterodimers in cis and in trans, as well as binding to other molecules, appear critical for their functions. Shedding by metalloproteases generates soluble factors interacting with cellular receptors and activating signal transduction.
- IgLON5
- psychiatric disorders
1. Introduction
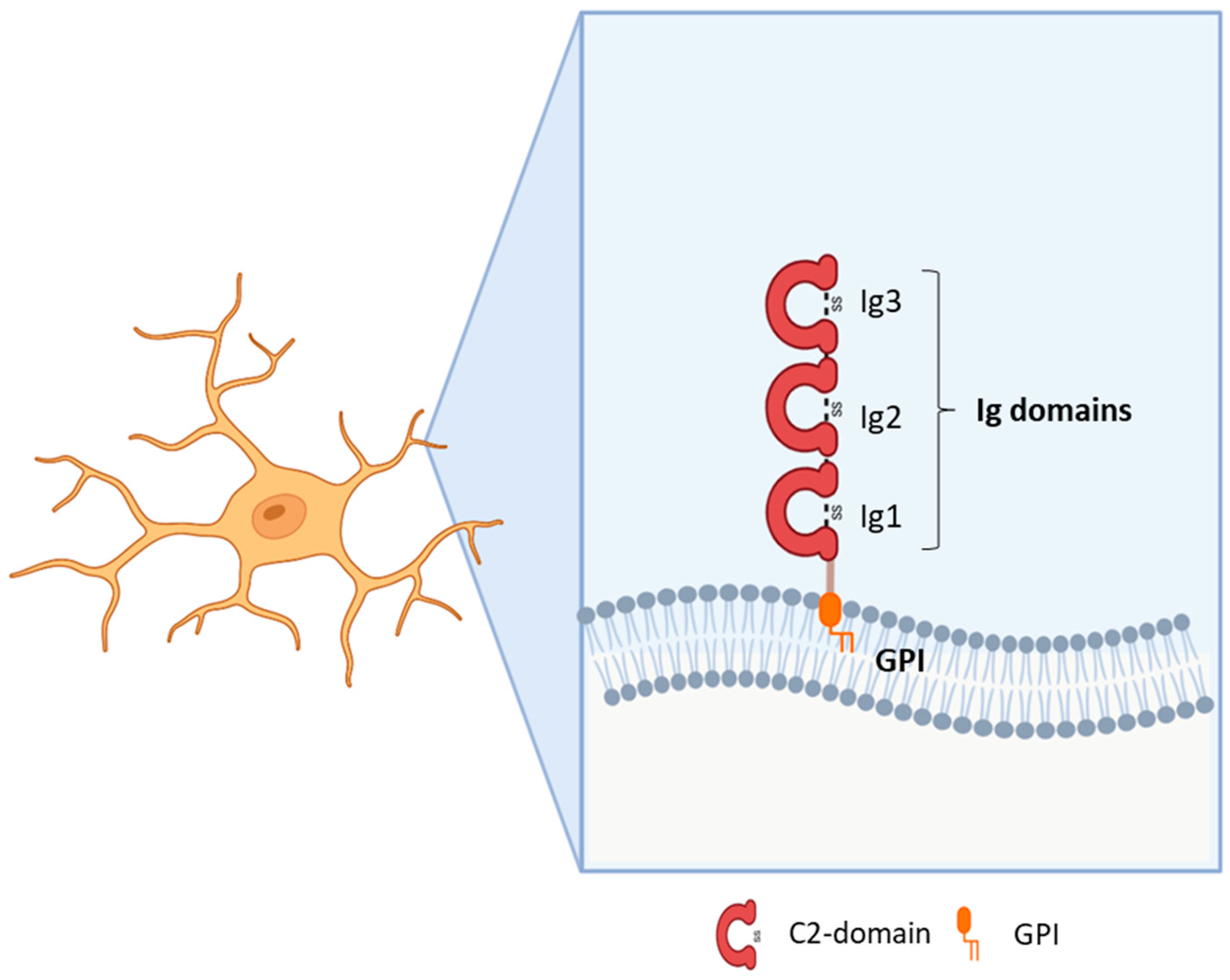
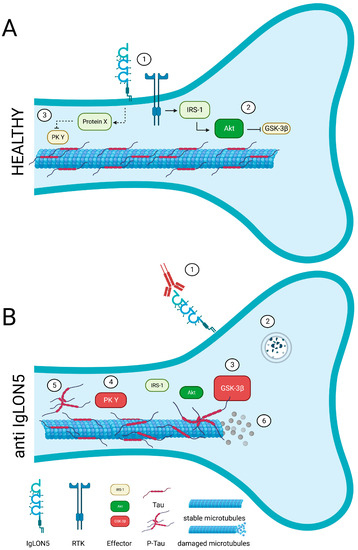
2. IgLON5 (IgLON Family Member 5)
References
- Shapiro, L.; Love, J.; Colman, D.R. Adhesion Molecules in the Nervous System: Structural Insights into Function and Diversity. Annu. Rev. Neurosci. 2007, 30, 451–474.
- Stachowicz, K. Physicochemical Principles of Adhesion Mechanisms in the Brain. Int. J. Mol. Sci. 2023, 24, 5070.
- Kadry, Y.A.; Calderwood, D.A. Chapter 22: Structural and signaling functions of integrins. Biochim. Biophys. Acta—Biomembr. 2020, 1862, 183206.
- Hynes, R.O. Integrins: Bidirectional, allosteric signaling machines. Cell 2002, 110, 673–687.
- McEver, R.P.; Zhu, C. Rolling cell adhesion. Annu. Rev. Cell Dev. Biol. 2010, 26, 363–396.
- Ley, K.; Kansas, G.S. Selectins in T-cell recruitment to non-lymphoid tissues and sites of inflammation. Nat. Rev. Immunol. 2004, 4, 325–335.
- Siew, J.J.; Chern, Y. Microglial Lectins in Health and Neurological Diseases. Front. Mol. Neurosci. 2018, 11, 158.
- Angiari, S. Selectin-mediated leukocyte trafficking during the development of autoimmune disease. Autoimmun. Rev. 2015, 14, 984–995.
- Redies, C. Cadherins in the central nervous system. Prog. Neurobiol. 2000, 61, 611–648.
- Takeichi, M. The cadherin superfamily in neuronal connections and interactions. Nat. Rev. Neurosci. 2007, 8, 11–20.
- Cameron, S.; McAllister, A.K. Immunoglobulin-Like Receptors and Their Impact on Wiring of Brain Synapses. Annu. Rev. Genet. 2018, 52, 567–590.
- Barclay, A.N. Membrane proteins with immunoglobulin-like domains—A master superfamily of interaction molecules. Semin. Immunol. 2003, 15, 215–223.
- Chothia, C.; Gelfand, I.; Kister, A. Structural determinants in the sequences of immunoglobulin variable domain. J. Mol. Biol. 1998, 278, 457–479.
- Walsh, F.S.; Doherty, P. Neural cell adhesion molecules of the immunoglobulin superfamily: Role in axon growth and guidance. Annu. Rev. Cell Dev. Biol. 1997, 13, 425–456.
- Sakurai, T. The role of cell adhesion molecules in brain wiring and neuropsychiatric disorders. Mol. Cell. Neurosci. 2017, 81, 4–11.
- Zinn, K.; Özkan, E. Neural immunoglobulin superfamily interaction networks. Curr. Opin. Neurobiol. 2017, 45, 99–105.
- Tan, R.P.A.; Leshchyns’ka, I.; Sytnyk, V. Glycosylphosphatidylinositol-Anchored Immunoglobulin Superfamily Cell Adhesion Molecules and Their Role in Neuronal Development and Synapse Regulation. Front. Mol. Neurosci. 2017, 10, 378.
- Leshchyns’ka, I.; Sytnyk, V. Reciprocal Interactions between Cell Adhesion Molecules of the Immunoglobulin Superfamily and the Cytoskeleton in Neurons. Front. Cell Dev. Biol. 2016, 4, 9.
- Maness, P.F.; Schachner, M. Neural recognition molecules of the immunoglobulin superfamily: Signaling transducers of axon guidance and neuronal migration. Nat. Neurosci. 2007, 10, 19–26.
- Karagogeos, D. Neural GPI-anchored cell adhesion molecules. Front. Biosci. 2003, 8, s1304–s1320.
- Levitt, P. A monoclonal antibody to limbic system neurons. Science 1984, 223, 299–301.
- Schofield, P.R.; McFarland, K.C.; Hayflick, J.S.; Wilcox, J.N.; Cho, T.M.; Roy, S.; Lee, N.M.; Loh, H.H.; Seeburg, P.H. Molecular characterization of a new immunoglobulin superfamily protein with potential roles in opioid binding and cell contact. EMBO J. 1989, 8, 489–495.
- Struyk, A.F.; Canoll, P.D.; Wolfgang, M.J.; Rosen, C.L.; D’Eustachio, P.; Salzer, J.L. Cloning of neurotrimin defines a new subfamily of differentially expressed neural cell adhesion molecules. J. Neurosci. 1995, 15, 2141–2156.
- Funatsu, N.; Miyata, S.; Kumanogoh, H.; Shigeta, M.; Hamada, K.; Endo, Y.; Sokawa, Y.; Maekawa, S. Characterization of a novel rat brain glycosylphosphatidylinositol- anchored protein (Kilon), a member of the IgLON cell adhesion molecule family. J. Biol. Chem. 1999, 274, 8224–8230.
- Marg, A.; Sirim, P.; Spaltmann, F.; Plagge, A.; Kauselmann, G.; Buck, F.; Rathjen, F.G.; Brümmendorf, T. Neurotractin, a novel neurite outgrowth-promoting Ig-like protein that interacts with CEPU-1 and LAMP. J. Cell Biol. 1999, 145, 865–876.
- Sabater, L.; Gaig, C.; Gelpi, E.; Bataller, L.; Lewerenz, J.; Torres-Vega, E.; Contreras, A.; Giometto, B.; Compta, Y.; Embid, C.; et al. A novel non-rapid-eye movement and rapid-eye-movement parasomnia with sleep breathing disorder associated with antibodies to IgLON5: A case series, characterisation of the antigen, and post-mortem study. Lancet Neurol. 2014, 13, 575–586.
- Takamori, S.; Holt, M.; Stenius, K.; Lemke, E.A.; Grønborg, M.; Riedel, D.; Urlaub, H.; Schenck, S.; Brügger, B.; Ringler, P.; et al. Molecular anatomy of a trafficking organelle. Cell 2006, 127, 831–846.
- Pischedda, F.; Piccoli, G. The IgLON Family Member Negr1 Promotes Neuronal Arborization Acting as Soluble Factor via FGFR2. Front. Mol. Neurosci. 2016, 8, 89.
- Szczurkowska, J.; Pischedda, F.; Pinto, B.; Managò, F.; Haas, C.A.; Summa, M.; Bertorelli, R.; Papaleo, F.; Schäfer, M.K.; Piccoli, G.; et al. NEGR1 and FGFR2 cooperatively regulate cortical development and core behaviours related to autism disorders in mice. Brain 2018, 141, 2772–2794.
- Fearnley, S.; Raja, R.; Cloutier, J.F. Spatiotemporal expression of IgLON family members in the developing mouse nervous system. Sci. Rep. 2021, 11, 19536.
- Kubick, N.; Brösamle, D.; Mickael, M.E. Molecular Evolution and Functional Divergence of the IgLON Family. Evol. Bioinforma. 2018, 14, 1176934318775081.
- Leshchyns’ka, I.; Sytnyk, V. Synaptic Cell Adhesion Molecules in Alzheimer’s Disease. Neural Plast. 2016, 2016, 6427537.
- Wennström, M.; Nielsen, H.M. Cell adhesion molecules in Alzheimer’s disease. Degener. Neurol. Neuromuscul. Dis. 2012, 2, 65–77.
- Leshchyns’ka, I.; Liew, H.T.; Shepherd, C.; Halliday, G.M.; Stevens, C.H.; Ke, Y.D.; Ittner, L.M.; Sytnyk, V. Aβ-dependent reduction of NCAM2-mediated synaptic adhesion contributes to synapse loss in Alzheimer’s disease. Nat. Commun. 2015, 6, 8836.
- Tang, X.; Tena, J.; Di Lucente, J.; Maezawa, I.; Harvey, D.J.; Jin, L.-W.; Lebrilla, C.B.; Zivkovic, A.M. Transcriptomic and glycomic analyses highlight pathway-specific glycosylation alterations unique to Alzheimer’s disease. Sci. Rep. 2023, 13, 7816.
- Sytnyk, V.; Leshchyns’ka, I.; Schachner, M. Neural Cell Adhesion Molecules of the Immunoglobulin Superfamily Regulate Synapse Formation, Maintenance, and Function. Trends Neurosci. 2017, 40, 295–308.
- Gaig, C.; Graus, F.; Compta, Y.; Högl, B.; Bataller, L.; Brüggemann, N.; Giordana, C.; Heidbreder, A.; Kotschet, K.; Lewerenz, J.; et al. Clinical manifestations of the anti-IgLON5 disease. Neurology 2017, 88, 1736–1743.
- Nissen, M.S.; Blaabjerg, M. Anti-IgLON5 Disease: A Case With 11-Year Clinical Course and Review of the Literature. Front. Neurol. 2019, 10, 1056.
- Werner, J.; Jelcic, I.; Schwarz, E.I.; Probst-Müller, E.; Nilsson, J.; Schwizer, B.; Bloch, K.E.; Lutterotti, A.; Jung, H.-H.; Schreiner, B. Anti-IgLON5 Disease: A New Bulbar-Onset Motor Neuron Mimic Syndrome. Neurol.-Neuroimmunol. Neuroinflamm. 2021, 8, e962.
- Gelpi, E.; Höftberger, R.; Graus, F.; Ling, H.; Holton, J.L.; Dawson, T.; Popovic, M.; Pretnar-Oblak, J.; Högl, B.; Schmutzhard, E.; et al. Neuropathological criteria of anti-IgLON5-related tauopathy. Acta Neuropathol. 2016, 132, 531–543.
- Ranaivoson, F.M.; Turk, L.S.; Ozgul, S.; Kakehi, S.; von Daake, S.; Lopez, N.; Trobiani, L.; De Jaco, A.; Denissova, N.; Demeler, B.; et al. A Proteomic Screen of Neuronal Cell-Surface Molecules Reveals IgLONs as Structurally Conserved Interaction Modules at the Synapse. Structure 2019, 27, 893–906.e9.
- Sabater, L.; Planagumà, J.; Dalmau, J.; Graus, F. Cellular investigations with human antibodies associated with the anti-IgLON5 syndrome. J. Neuroinflamm. 2016, 13, 226.
- Landa, J.; Gaig, C.; Plagumà, J.; Saiz, A.; Antonell, A.; Sanchez-Valle, R.; Dalmau, J.; Graus, F.; Sabater, L. Effects of IgLON5 Antibodies on Neuronal Cytoskeleton: A Link between Autoimmunity and Neurodegeneration. Ann. Neurol. 2020, 88, 1023–1027.
- Ryding, M.; Gamre, M.; Nissen, M.S.; Nilsson, A.C.; Okarmus, J.; Poulsen, A.A.E.; Meyer, M.; Blaabjerg, M. Neurodegeneration Induced by Anti-IgLON5 Antibodies Studied in Induced Pluripotent Stem Cell-Derived Human Neurons. Cells 2021, 10, 837.
- Honorat, J.A.; Komorowski, L.; Josephs, K.A.; Fechner, K.; St Louis, E.K.; Hinson, S.R.; Lederer, S.; Kumar, N.; Gadoth, A.; Lennon, V.A.; et al. IgLON5 antibody: Neurological accompaniments and outcomes in 20 patients. Neurol.-Neuroimmunol. Neuroinflamm. 2017, 4, e385.
- Erro, M.E.; Sabater, L.; Martínez, L.; Herrera, M.; Ostolaza, A.; García de Gurtubay, I.; Tuñón, T.; Graus, F.; Gelpi, E. Anti-IGLON5 disease: A new case without neuropathologic evidence of brainstem tauopathy. Neurol.-Neuroimmunol. Neuroinflamm. 2020, 7, e651.
- Xia, Y.; Prokop, S.; Giasson, B.I. “Don’t Phos Over Tau”: Recent developments in clinical biomarkers and therapies targeting tau phosphorylation in Alzheimer’s disease and other tauopathies. Mol. Neurodegener. 2021, 16, 37.
- Hernandez, F.; Lucas, J.J.; Avila, J. GSK3 and tau: Two convergence points in Alzheimer’s disease. J. Alzheimer’s Dis. 2013, 33 (Suppl. 1), S141–S144.
- Kanno, T.; Tsuchiya, A.; Tanaka, A.; Nishizaki, T. Combination of PKCε Activation and PTP1B Inhibition Effectively Suppresses Aβ-Induced GSK-3β Activation and Tau Phosphorylation. Mol. Neurobiol. 2016, 53, 4787–4797.
- Landa, J.; Serafim, A.B.; Gaig, C.; Saiz, A.; Koneczny, I.; Hoftberger, R.; Santamaria, J.; Dalmau, J.; Graus, F.; Sabater, L. Patients’ IgLON5 autoantibodies interfere with IgLON5-protein interactions. Front. Immunol. 2023, 14, 1151574.