Oxidative stress is increasingly recognized as a central player in a range of gastrointestinal (GI) disorders, as well as complications stemming from therapeutic interventions. The dysfunction of the ENS is characteristic of a spectrum of disorders, including neurointestinal diseases and conditions such as inflammatory bowel disease (IBD), diabetic gastroparesis, and chemotherapy-induced GI side effects. Neurons in the Enteric nervous system (ENS), while essential for normal gut function, appear particularly vulnerable to oxidative damage. Mechanistically, oxidative stress in enteric neurons can result from intrinsic nitrosative injury, mitochondrial dysfunction, or inflammation-related pathways. Although antioxidant-based therapies have shown limited efficacy, recognizing the multifaceted role of oxidative stress in GI diseases offers a promising avenue for future interventions.
- oxidative stress
- reactive oxygen species
- gastrointestinal
- enteric nervous system
- enteric neuron
1. Introduction
2. Oxidative Stress
Cellular organisms harness oxygen’s reactivity to generate substantial energy, vital for sustaining the intricate multicellular lifeforms existing today. This oxidative potential, while advantageous for cellular metabolism and oxidative phosphorylation, also poses risks to cellular structures, necessitating antioxidant defense mechanisms for survival [1,2][1][2]. The resultant dualistic nature of oxygen and its reactive oxygen species (ROS) derivatives has rendered reduction-oxidative (redox) equilibrium pivotal in regulating diverse cellular processes [3,4][3][4]. Although ROS and reactive nitrogen species (RNS) are commonly acknowledged as agents of cellular impairment, they are concurrently integrated into normative physiological functions. At moderate levels, they mediate apoptosis, intracellular signaling cascades, transcriptional processes, oxygen sensing, and smooth muscle tone [2]. The immune system harnesses ROS for bactericidal properties via enzymatic reactions by leukocytes [5,6,7][5][6][7]. This becomes notably germane within the GI tract, which accommodates approximately 100 trillion commensal bacteria in humans. Nitric oxide (NO), generated enzymatically by three distinct nitric oxide synthase (NOS) isoforms, exemplifies a molecule deeply intertwined with diverse physiological functions. Disruption of this delicate redox balance towards a pro-oxidative milieu culminates in oxidative stress whereby ROS, directly or indirectly, inflict structural alterations to lipids, proteins, and DNA, triggering cellular damage and inflammation [9,10][8][9]. The genesis of oxidative stress originates from either a perturbation in ROS/RNS production or their neutralization by antioxidants. Nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (NOX) and xanthine oxidase (XOD) are the primary sources of superoxide (O2.−) radicals [11][10]. Other notable ROS and RNS include the hydroxyl radical (•OH), hydrogen peroxide (H2O2), hypochlorous acid (HOCl), nitric oxide (NO), and peroxynitrite (ONOO-), which fulfill crucial roles in immune responses, albeit excessive production leads to cellular dysfunction and/or cell death [4]. Immune cells generate ROS/RNS via enzymes such as NOX, inducible nitric oxide synthase (iNOS), and myeloperoxidase (MPO), which are pivotal in the normal immune response yet deleterious under prolonged inflammation [5,12,13][5][11][12]. Moreover, excessive O2.− produced within the electron transport chain (ETC) complexes of the mitochondria can inflict damage and induce programmed cell death, especially when reacting with NO to form ONOO- [14,15][13][14]. Mitochondrial detoxification of O2.− to H2O2 is facilitated by superoxide dismutase (SOD) [13][12]. However, H2O2 may also emanate from diverse metabolic processes and dual oxidases (DUOX) [16][15]. While relatively more stable than O2.−, the susceptibility of H2O2 to react with metals, such as Fe2+ via the Fenton reaction, renders its detoxification by catalase (CAT) imperative [17][16]. The glutathione system, encompassing glutathione peroxidase and glutathione reductase, further amplifies the cellular arsenal against excessive ROS accumulation [18][17]. The gut’s heme oxygenase-1 (HO-1) offers antioxidant potential through heme catabolism and the production of CO, ferritin, and bilirubin [19,20,21,22][18][19][20][21]. Evidently, oxidative stress emerges as a central protagonist in an array of acquired and congenital gastrointestinal disorders, as well as a mediator of adverse effects from therapeutic interventions and procedures.2.1. Oxidative Stress in GI Disease
2.1.1. Drug/Toxin Exposure
Chemotherapeutic agents initially produce high levels of ROS, which are a primary cause of pathology following the use of agents such as anthracyclines (daunorubicin, doxorubicin), platinum-based compounds (cisplatin, carboplatin, oxaliplatin), epipodophyllotoxins (e.g., etoposide and tiliroside), alkylating agents, and camptothecins [84][22]. In addition, the administration of mitoxantrone, actinomycin D, enediynes such as bleomycin, elasmin A, chartreusins, 5-fluorouracil (5-FU), and irinotecan can induce severe oxidative stress [85,86,87][23][24][25]. It is widely considered that chemotherapeutic agents depend largely on ROS generation to destroy cancerous cells [88][26]. However, ROS also contribute to many common side effects, including GI toxicity and mutagenesis [85,89][23][27]. The deleterious effects of the overproduction of ROS include severe mucosal damage [31[22][28],84], loss of epithelial cells and tight junction proteins [90][29], microbiota imbalance [91[30][31],92], and enteric neuropathy [30][32]. Nearly all chemotherapy patients suffer from GI side effects, such as nausea, vomiting, diarrhea, constipation, and ulceration [30,93][32][33]. Mucositis is one of the most undesired side effects of antineoplastic chemotherapeutics, presenting as severe inflammation of the GI mucosa [96][34]. Chemotherapy-induced mucositis is responsible for poor clinical outcomes, including an increased risk of infection, prolonged hospitalization, and even death [97][35]. Mucositis is associated with various symptoms, such as nausea, severe diarrhea, GI bleeding, and severe abdominal pain [98][36]. It is well established that the pathogenesis of mucositis correlates with the overwhelming production of ROS and inflammatory mediators [99][37]. The primary mediator of mucosal damage after chemotherapy is the overproduction of ROS [98][36]. Oxidative stress leads to DNA damage in epithelial progenitor cells, increased production of inflammatory mediators, cellular apoptosis, and a progressive loss of cells from the absorptive surface. Radiotherapy is an important treatment modality for abdominal and pelvic malignancies; however, GI complications and enteropathy are common sequelae after exposure. Oxidative stress is considered the driving force through which radiation induces cell death in neoplastic cells [101][38]. Radiotherapy generates a large number of free radicals, which are predominantly formed by the radiolysis of water to •OH but can also be produced by the mitochondria [102][39]. These free radicals target nuclear DNA and cell structures to induce cell death in rapidly proliferating cells, leading to inevitable off-target effects on healthy cells. In models of radiation injury, irradiation results in an increase in the lipid peroxidation product malondialdehyde (MDA), indicative of oxidative stress in the small bowel [54][40]. Nonsteroidal anti-inflammatory drugs (NSAIDs) are generally considered safe and are widely used in the clinical setting for their analgesic and anti-platelet properties. Nevertheless, there is a clear risk of developing GI complications, including gastroduodenal ulcers, with prolonged NSAID usage. The metabolites of NSAIDs can have pro-oxidant properties [103][41] or induce the generation of ROS in other cells. For example, indomethacin can increase mitochondrial O2.− and XOD expression directly in colonic epithelial cells in in vitro models, resulting in increased O2.− generation [104,105][42][43]. Oxidative stress is recognized as critical to the pathogenesis of gastroduodenal ulcers. Early evidence of this association included observations of the depletion of antioxidant enzymes, such as SOD, in gastric and duodenal biopsies [46][44]. Chronic alcohol consumption is also associated with GI toxicity, and chronic intestinal pseudo-obstruction has previously been linked to fetal alcohol syndrome [106][45]. Alcohol consumption results in oxidative stress via the oxidative byproducts of ethanol metabolism and nicotinamide adenine dinucleotide (NAD) depletion. In models of chronic alcohol exposure, protein nitration has been associated with damage to the intestinal barrier, increasing its permeability and implicating NO in its pathogenesis, notably prior to any evidence of liver disease [34][46]. Beyond the liver, enzymes responsible for ethanol metabolism can be found in the gut, including high levels of cytochrome P450 2E1 (CYP2E1), which generates free radicals that were shown to trigger oxidative stress-dependent changes in epithelial barrier permeability in in vitro models [35][47].2.1.2. Ischemia–Reperfusion and Postoperative Injury
Ischemia–reperfusion injury is an important clinical problem for ischemic syndromes and solid organ transplantation and occurs in several tissues upon reoxygenation. The mechanisms involved are largely considered to be driven by oxidative stress followed by the activation of an immune response in the injured tissue [107][48]. The gut is considered to be highly susceptible to ischemia–reperfusion injury due to its ability to generate a large number of free radicals. Xanthine dehydrogenase and XOD are interconvertible enzymes from the same gene product [108][49]. During intestinal ischemia, xanthine dehydrogenase is converted to XOD, which is capable of producing the free radicals O2·− and H2O2 from oxygen. In the ischemic environment deprived of oxygen, this is of little consequence; however, reoxygenation results in a rapid influx of the oxygen substrate, which is subsequently converted to a burst of free radicals, causing oxidative stress [43,109][50][51]. Tissue ischemia resulting in bowel injury can be caused by several conditions and often requires surgical resection to remove the affected tissue. Given that ischemia–reperfusion is associated with oxidative stress, this process was modeled in rats to evaluate the effects of hypoxia by portal vein occlusion on bowel anastomotic healing. In this study, oxidative stress, as measured by lipid peroxidation and protein oxidation, was associated with poor anastomotic healing and a lack of collagen deposition [26][52]. Nevertheless, ischemia is not necessarily required to induce oxidative stress in bowel anastomoses. The physical bowel injury from surgery alone also appears to evoke oxidative stress in intestinal tissues. Postoperative ileus, or paralytic ileus, is a common condition in which the peristaltic activity of the intestine is diminished after bowel manipulation during surgical procedures. Many studies have investigated the mechanisms of postoperative ileus, revealing a complex condition that involves an initial activation of cells in the muscularis propria [110][53]. These cells may include resident macrophages, mast cells, enteric glia, and enteric neurons and potentially an influx of infiltrating immune cells, such as monocytes and neutrophils, at later stages that disrupt normal intestinal motility [110,111][53][54]. Notably, in a mouse model of postoperative ileus, oxidative stress was indicated by elevated lipid peroxidation levels as early as the first postoperative hour [78][55]. MPO and iNOS are two of the largest sources of immune system-derived ROS/RNS and were upregulated in this model; however, this did not occur until 3 h postoperatively and coincided with the onset of inflammatory cytokine production, including interleukin-6 (IL-6) and monocyte chemoattractant protein-1 (MCP-1) [78][55]. This suggests that oxidative stress may occur independent of inflammation in the immediate stage following tissue injury and instead may have a role in promoting the inflammatory response through the rapid oxidative burst.2.1.3. Congenital Disorders
Genetic GI conditions may also be primarily associated with oxidative stress, though due to the complexity of mutations, more research in this space is required. One example is Triple-A syndrome, whereby esophageal achalasia presents as a key feature. In this disease, mutation of the AAAS gene likely results in disturbed redox homeostasis, as demonstrated by oxidized and reduced glutathione balances after the knockdown of AAAS in human adrenocortical tumor and neuroblastoma cells [57][56]. Subsequently, these cells are highly susceptible to cell death following oxidative insult [57][56]. Other conditions associated with oxidative stress could include forms of chronic intestinal pseudo-obstruction that are associated with mutations in mitochondrial genes and mitochondrial neurogastrointestinal encephalopathy (MNGIE). Given that the mitochondria are the largest source of intracellular ROS, damage to the mitochondria could cause the uncoupling of oxidative phosphorylation and hampering of detoxifying mechanisms, which could contribute to the underlying pathophysiology of these diseases [113][57]. Infantile hypertrophic pyloric stenosis is another multifactorial genetic disease that can cause debilitating obstruction. While the etiology of this condition is unclear, it has been associated with a lack of NO production and can be modeled in the Hph-1 knock-out mouse model, which lacks tetrahydrobiopterin (BH4), an important cofactor for NO synthesis [80][58]. In this model, nNOS was upregulated as a possible compensatory mechanism; however, elevated levels of H2O2 and O2.− are suggestive of possible nNOS uncoupling [80][58].2.1.4. Inflammation and Infection
Oxidative stress constitutes an essential part of normal immune system functions, and free radicals are utilized as a delicate tool to eliminate bacteria and unhealthy cells and to modify the cellular response by inducing inflammatory transcriptional pathways and altering protein function. Nevertheless, free radical signaling acts in a non-specific manner, which can result in unwanted tissue damage reciprocally driving inflammatory processes. This has been best studied in inflammatory bowel disease (IBD) within GI research. IBD collectively encompasses UC and Crohn’s disease (CD), two incurable conditions in which patients experience periods of remission, flares, and relapses of inflammation within the GI tract that ultimately requires bowel resection in up to 80% of cases [115,116,117][59][60][61]. In UC, inflammation is observed in the colon and rectum, whilst in CD, the entire GI tract, but predominantly the colon and terminal ileum, may be inflamed [115,116][59][60]. Oxidative stress is prominent in IBD patients and experimental models of colitis [73,75,118,119][62][63][64][65]. A decline in the scavenging of free radicals is reported in IBD patients [71,73][62][66]. Necrotizing enterocolitis (NEC) is a life-threatening GI disease affecting premature and low birthweight infants. NEC also has important links to tissue oxygenation levels in the pathogenesis of the disease. This condition is considered multifactorial and involves an element of immune stimulation in the premature intestine during colonization with intestinal flora [122][67]. Total oxidant status (TOS) and the oxidative stress index (OSI) are increased in the serum of patients with NEC compared to preterm healthy controls, which further correlate with disease severity [59][68]. Notably, markers of oxidative stress, including advanced oxidation protein products and total hydroperoxides, in cord blood are predictive of developing NEC, which suggests that disturbances in the redox balance may precede disease onset [60][69]. After birth, there is a dramatic shift in oxygenation from the intra to the extrauterine environment. At later stages of gestation, antioxidant defenses are strengthened prior to birth [58][70]. Oxidative stress has also been implicated in parasitic infections of the intestine. Chagas disease is caused by the Trypanosoma cruzi (T. cruzi) parasite infection, but megacolon or megaesophagus occurs only in a subset of patients. Recently, the MRPS18B P260A gene variant was identified in 38.4% of patients with Chagas megaesophagus compared to 2.2% of the asymptomatic population [64][71]. To ascertain the potential function of the gene variant, the authors generated the Epstein–Barr virus-immortalized B lymphoblastoid cell lines from patients and showed that stimulation of these cells with the cytokine interferon-γ resulted in increased protein nitration and O2.− generation from the mitochondria, implicating nitro-oxidative stress as a potential mechanism.2.1.5. Cancer
It is established that intestinal inflammation causes oxidative stress which results in a high burden of free radicals secondary to the pathology, but inflammation also greatly enhances the risk of carcinogenesis, and oxidative stress may be considered a key instigator. In IBD, there is a substantial increase in the risk of developing colorectal cancer. Additionally, there is an increased risk of other malignancies, including small bowel cancer and extra-intestinal cancers, all of which have been linked to chronic oxidative stress-induced mutagenesis [124,125,126][72][73][74]. Barrett’s esophagus similarly involves oxidative stress, ultimately contributing to the development of esophageal adenocarcinoma [127][75]. In Barrett’s esophagus, the squamous mucosa of the distal esophagus is replaced by metaplastic columnar epithelium after prolonged insult, predominantly by the reflux of gastric acid and bile salts in gastroesophageal reflux disease. In biopsies of Barrett’s esophagus, there is an increase in levels of peroxynitrite, O2.−, and GSH, indicating oxidative stress [23][76]. While oxidative stress mediates the therapeutic effects of radiotherapy and chemotherapy in the treatment of cancer, oxidative stress is also a major contributor to carcinogenesis itself. In colorectal cancer, free radicals derived from environmental sources, diet, sedentary lifestyle, and inflammation can lead to oxidative stress and cause genomic instability and mutagenesis, resulting in the transformation of healthy colonocytes to dysplastic and neoplastic cells [126][74]. Biopsies of primary colorectal tumors support oxidative stress-dependent mechanisms of genomic instability, given the presence of oxidized DNA adducts and increased lipid peroxidation products, MDA and 4-hydroxynonenal (4-HNE), both of which positively correlate with histological grade and clinical stage [38][77].2.1.6. Diabetes Mellitus
A large body of research exists regarding the role of oxidative stress in tissue injury from diabetes mellitus. Oxidative stress may be largely considered a secondary driver of the disease pathophysiology as the primary effects appear to be mediated by advanced glycation end product (AGE) signaling and low-grade inflammation. In the GI tract, diabetes is associated with the development of gastroparesis and intestinal dysmotility. In the non-obese diabetic mouse model of gastroparesis, only a portion of mice develop delayed gastric emptying [19,20][18][19]. In the mice that exhibit this phenotype, elevated lipid peroxidation (as indicated by MDA levels) is observed compared to those with normal gastric emptying. Loss of nNOS (nitrergic neurons) and c-KIT (interstitial cells of Cajal, ICC) is observed, which normally mediate the relaxation of the pyloric sphincter to allow transit of gastric contents. Interestingly, the mice that do not develop delayed gastric emptying exhibit higher expression of the antioxidant enzyme HO-1, which was later shown to be expressed by gastric macrophages and may serve to protect cells of the muscularis propria from oxidative stress [19,20][18][19]. Oxidative stress has also been implicated in the streptozotocin (STZ) model of diabetes, with reports of elevated lipid peroxidation and protein oxidation after 6 weeks [66][78].3. Impact of Oxidative Stress on the Enteric Nervous System and Associated Sequelae
The ENS, an integral component of the autonomic nervous system, intricately regulates the physiological functions of the GI tract by orchestrating effector systems like musculature, secretion, and vasculature. This distinctive influence has positioned the ENS as an alluring and innovative therapeutic target for a spectrum of GI disorders [93,131][33][79]. Structurally, this intricate neural network comprises two distinct plexuses—the outer myenteric plexus and the inner submucosal plexus [132][80]. The myenteric plexus is characterized by its location between the outer longitudinal and inner circular smooth muscle layers of the intestinal wall, where enteric neurons control the rhythm and coordination of muscular contractions and engage with tissue-resident muscularis macrophages [133][81]. Dysfunction of the ENS is a hallmark of several gastrointestinal disorders, encompassing alterations in its structure, neuroinflammatory responses, aberrations in neuronal excitability and signaling, and conditions such as enteric neuropathy or aganglionosis. This disruption leads to imbalances in gut homeostasis, often culminating in dysmotility or aperistalsis. This constitutes a spectrum of disorders comprised of idiopathic gastroparesis, Hirschsprung’s disease, esophageal achalasia, Chagas disease, pyloric stenosis, mitochondrial neurogastrointestinal encephalomyopathy (MNGIE), and certain manifestations of chronic intestinal pseudo-obstruction, collectively termed as neurointestinal diseases, which underscore the profound impact of ENS aberrations on gut physiology [135][82]. Conditions such as IBD, diabetic gastroparesis, and chemotherapy-induced GI complications, even though triggered by different pathogenesis, exhibit comparable disruptions in neurally mediated functions, contributing to significant morbidity. Interestingly, preclinical studies in models of Parkinson’s disease have demonstrated enteric neuroinflammation in the early stages of disease pathogenesis prior to the onset of neurological symptoms, suggesting that the deleterious effects of enteric oxidative stress may also be pertinent as a biomarker or a part of the pathophysiology of disorders affecting the central nervous system (CNS) [136,137][83][84]. The consequences of ENS aberrations are far-reaching, inducing a spectrum of debilitating sequelae ranging from profound dysmotility to potentially fatal complications like impaction or perforation. The restoration of ENS integrity emerges as a paramount objective not only to enhance quality of life but also to avert these grave complications. Intriguingly, a common thread among several acquired GI disorders involving ENS dysfunction is their association with oxidative stress. This intricately woven relationship between neuronal damage and oxidative stress remains an active area of investigation, seeking to illuminate the mechanistic underpinnings and potential therapeutic avenues to mitigate these debilitating conditions.3.1. Antioxidant Defense in Enteric Neurons
In general, neurons are particularly susceptible to oxidative insult from free radicals due to their higher energy demand and O2 consumption, excess mitochondria-derived O2.−, auto-oxidation of neurotransmitters, excitotoxicity, poor antioxidant defenses, and limited replicative potential [138][85]. Neurons of the ENS are also highly sensitive to oxidative stress, which has been shown to alter their electrophysiological properties, damage neuronal membranes, and trigger neuronal death [69,139,140][86][87][88]. The role of oxidative stress in neuronal damage was shown in models of chemotherapy, diabetes, physiological aging, and Chagas disease [30,53,65,68,141][32][89][90][91][92]. Similarly, oxidative stress is predicted to be a key contributor to ENS dysfunction in the pathophysiology of IBD [118,131][64][79]. In physiological aging of the intestine, enteric neuron loss was observed in mice after 17 months and associated with increased ROS and markers of apoptosis [53][89].3.2. Mechanism of Enteric Neuropathy Involving Oxidative Stress
Currently, the known mechanisms of oxidative stress in enteric neurons can be categorized as related to either intrinsic nitrosative injury, mitochondrial dysfunction, or inflammation-related oxidative stress (Figure 1). However, these pathways may not always be mutually exclusive. Intrinsic nitrosative injury can often be identified by excessive loss of nNOS neurons; however, in intestinal inflammation and chemotherapy exposure, nNOS-expressing neurons are not specifically lost [119,144,145,146,147,148,149,150,151][65][93][94][95][96][97][98][99][100]; therefore, non-nitrosative stress mechanisms of enteric neuropathy are more likely in these conditions.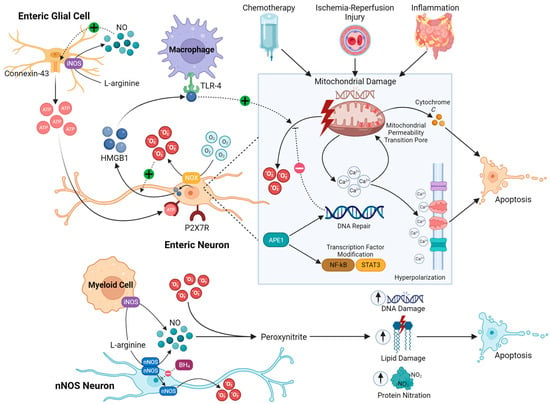
3.2.1. Intrinsic Nitrosative Injury
3.2.2. Mitochondrial Dysfunction
Myenteric neurons appear to exhibit the highest density of mitochondria, which may explain their susceptibility to increases in O2.− production in pathological conditions [30,156][32][106]. Local oxygen gradients could also affect ROS generation by the mitochondria and the redox balance. Excessive ROS are produced in a lower-energy oxidative environment or a higher-energy reductive environment [85,157][23][107]. A reductive environment is usually attributed to hypoxic conditions, and tissue hypoxia has been studied in depth in the brain as a cause of neuronal loss mediated by ROS released by damaged mitochondria [158][108]. Under physiological conditions, the kinetics of O2.− production by the mitochondria are directly proportional to the levels of O2; this has been demonstrated in neurons of the peripheral nervous system (PNS) and CNS [159,160,161,162][109][110][111][112]. Therefore, there is also potential for a hyperoxic environment to cause neuronal damage, as shown in cortical neurons [163][113]. Mitochondrial dysfunction has been strongly linked to enteric neuropathy caused by chemotherapy. Treatment with the platinum-based chemotherapeutic drug oxaliplatin induces the loss of enteric neurons, including nNOS-expressing neurons in the submucosal and myenteric plexuses. This correlated with severe colonic dysmotility and constipation [30][32]. The proportion of nNOS-immunoreactive neurons after oxaliplatin exposure was higher in the submucosal and myenteric plexuses of the distal colon, indicating that this is likely not the result of an intrinsic nitrergic neuropathy as observed in other GI conditions.3.2.3. Enteric Neuroinflammation
Oxidative stress and chronic neuroinflammation intertwine as key pathologic factors contributing to enteric neuropathy [131,149][79][98]. This is illustrated in chemically-induced colitis, which causes oxidative stress in the ENS and consequently dysfunction in neurally controlled intestinal functions [140][88]. In a model of parasitic ileitis, the greatest changes in lipid peroxidation were observed in the muscle layers rather than in the mucosa or plasma [169][114]. Therefore, the muscle layers of the intestine appear to be susceptible to oxidative injury. Likewise, myenteric neurons contained within the muscle layers are not resistant to oxidative stress induced by intestinal inflammation [69][86]. In intestinal inflammation, the role of oxidative stress in mediating neuronal cell death is exemplified by the administration of the antioxidant NAC, which attenuates neuronal loss in vivo in an acute model of dinitrobenzene sulfonic acid (DNBS)-induced colitis [69][86]. Additionally, NAC did not appear to directly ameliorate the inflammatory response, suggesting a greater contribution from oxidative insult than proinflammatory cytokines in driving neuropathy in colitis. Together, these data provide compelling evidence that oxidative stress is a critical player in GI disease, and subsequent damage to the ENS is likely a major cause of dysmotility that complicates and worsens disease course. This data suggests a novel therapeutic target for the management of multiple GI conditions; however, the clinical application of antioxidant compounds has been met with limited success, predominantly due to the inadequate scavenging properties of small molecule antioxidants and low bioavailability [3].References
- Fisher, A.B. Redox signaling across cell membranes. Antioxid. Redox Signal. 2009, 11, 1349–1356.
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84.
- Forman, H.J.; Zhang, H. Targeting oxidative stress in disease: Promise and limitations of antioxidant therapy. Nat. Rev. Drug Discov. 2021, 20, 689–709.
- Li, R.; Jia, Z.; Trush, M.A. Defining ROS in biology and medicine. React. Oxyg. Species 2016, 1, 9.
- Al Ghouleh, I.; Khoo, N.K.; Knaus, U.G.; Griendling, K.K.; Touyz, R.M.; Thannickal, V.J.; Barchowsky, A.; Nauseef, W.M.; Kelley, E.E.; Bauer, P.M.; et al. Oxidases and peroxidases in cardiovascular and lung disease: New concepts in reactive oxygen species signaling. Free Radic. Biol. Med. 2011, 51, 1271–1288.
- Rada, B.; Leto, T.L. Oxidative innate immune defenses by Nox/Duox family NADPH oxidases. Contrib. Microbiol. 2008, 15, 164–187.
- Soldati, T.; Neyrolles, O. Mycobacteria and the intraphagosomal environment: Take it with a pinch of salt(s)! Traffic 2012, 13, 1042–1052.
- Sies, H. Oxidative stress: A concept in redox biology and medicine. Redox Biol. 2015, 4, 180–183.
- Sies, H. Oxidative eustress: On constant alert for redox homeostasis. Redox Biol. 2021, 41, 101867.
- Poprac, P.; Jomova, K.; Simunkova, M.; Kollar, V.; Rhodes, C.J.; Valko, M. Targeting Free Radicals in Oxidative Stress-Related Human Diseases. Trends Pharmacol. Sci. 2017, 38, 592–607.
- Cho, K.J.; Seo, J.M.; Kim, J.H. Bioactive lipoxygenase metabolites stimulation of NADPH oxidases and reactive oxygen species. Mol. Cells 2011, 32, 1–5.
- Dan Dunn, J.; Alvarez, L.A.J.; Zhang, X.; Soldati, T. Reactive oxygen species and mitochondria: A nexus of cellular homeostasis. Redox Biol. 2015, 6, 472–485.
- Chen, Y.; Azad, M.B.; Gibson, S.B. Superoxide is the major reactive oxygen species regulating autophagy. Cell Death Differ. 2009, 16, 1040–1052.
- Ahmad, R.; Hussain, A.; Ahsan, H. Peroxynitrite: Cellular pathology and implications in autoimmunity. J. Immunoass. Immunochem. 2019, 40, 123–138.
- De Deken, X.; Corvilain, B.; Dumont, J.E.; Miot, F. Roles of DUOX-mediated hydrogen peroxide in metabolism, host defense, and signaling. Antioxid. Redox Signal. 2014, 20, 2776–2793.
- Winterbourn, C.C. Toxicity of iron and hydrogen peroxide: The Fenton reaction. Toxicol. Lett. 1995, 82, 969–974.
- Masella, R.; Di Benedetto, R.; Varì, R.; Filesi, C.; Giovannini, C. Novel mechanisms of natural antioxidant compounds in biological systems: Involvement of glutathione and glutathione-related enzymes. J. Nutr. Biochem. 2005, 16, 577–586.
- Choi, K.M.; Gibbons, S.J.; Nguyen, T.V.; Stoltz, G.J.; Lurken, M.S.; Ordog, T.; Szurszewski, J.H.; Farrugia, G. Heme oxygenase-1 protects interstitial cells of Cajal from oxidative stress and reverses diabetic gastroparesis. Gastroenterology 2008, 135, 2055–2064.e2.
- Choi, K.M.; Kashyap, P.C.; Dutta, N.; Stoltz, G.J.; Ordog, T.; Shea Donohue, T.; Bauer, A.J.; Linden, D.R.; Szurszewski, J.H.; Gibbons, S.J.; et al. CD206-positive M2 macrophages that express heme oxygenase-1 protect against diabetic gastroparesis in mice. Gastroenterology 2010, 138, 2399–2409.e1.
- Otterbein, L.E.; Choi, A.M. Heme oxygenase: Colors of defense against cellular stress. Am. J. Physiol. Lung Cell. Mol. Physiol. 2000, 279, L1029–L1037.
- Ryter, S.W.; Tyrrell, R.M. The heme synthesis and degradation pathways: Role in oxidant sensitivity. Heme oxygenase has both pro- and antioxidant properties. Free Radic. Biol. Med. 2000, 28, 289–309.
- Conklin, K.A. Chemotherapy-associated oxidative stress: Impact on chemotherapeutic effectiveness. Integr. Cancer Ther. 2004, 3, 294–300.
- Bhattacharyya, A.; Chattopadhyay, R.; Mitra, S.; Crowe, S.E. Oxidative stress: An essential factor in the pathogenesis of gastrointestinal mucosal diseases. Physiol. Rev. 2014, 94, 329–354.
- Orlikova, B.; Legrand, N.; Panning, J.; Dicato, M.; Diederich, M. Anti-inflammatory and anticancer drugs from nature. Cancer Treat. Res. 2014, 159, 123–143.
- Zeng, D.; Wang, Y.; Chen, Y.; Li, D.; Li, G.; Xiao, H.; Hou, J.; Wang, Z.; Hu, L.; Wang, L.; et al. Angelica Polysaccharide Antagonizes 5-FU-Induced Oxidative Stress Injury to Reduce Apoptosis in the Liver Through Nrf2 Pathway. Front. Oncol. 2021, 11, 720620.
- Yang, H.; Villani, R.M.; Wang, H.; Simpson, M.J.; Roberts, M.S.; Tang, M.; Liang, X. The role of cellular reactive oxygen species in cancer chemotherapy. J. Exp. Clin. Cancer Res. 2018, 37, 266.
- Conklin, K.A. Dietary antioxidants during cancer chemotherapy: Impact on chemotherapeutic effectiveness and development of side effects. Nutr. Cancer 2000, 37, 1–18.
- Rtibi, K.; Selmi, S.; Grami, D.; Sebai, H.; Amri, M.; Marzouki, L. Irinotecan chemotherapy-induced intestinal oxidative stress: Underlying causes of disturbed mucosal water and electrolyte transport. Pathophysiology 2017, 24, 275–279.
- Bai, G.W.; Han, D.Y.; Yang, Q.Y.; Xie, Y.; Guo, Z.X.; Zhou, W.L.; Deng, C.H.; Sun, X.Z. Oxidative stress induces damage to epididymal epithelial tight junction protein ZO-1 and impairs epididymal function in varicocele rats. Natl. J. Androl. 2019, 25, 302–308.
- Cao, S.S. Cellular Stress Responses and Gut Microbiota in Inflammatory Bowel Disease. Gastroenterol. Res. Pr. 2018, 2018, 7192646.
- Vivarelli, S.; Salemi, R.; Candido, S.; Falzone, L.; Santagati, M.; Stefani, S.; Torino, F.; Banna, G.L.; Tonini, G.; Libra, M. Gut Microbiota and Cancer: From Pathogenesis to Therapy. Cancers 2019, 11, 38.
- McQuade, R.M.; Carbone, S.E.; Stojanovska, V.; Rahman, A.; Gwynne, R.M.; Robinson, A.M.; Goodman, C.A.; Bornstein, J.C.; Nurgali, K. Role of oxidative stress in oxaliplatin-induced enteric neuropathy and colonic dysmotility in mice. Br. J. Pharmacol. 2016, 173, 3502–3521.
- McQuade, R.M.; Stojanovska, V.; Abalo, R.; Bornstein, J.C.; Nurgali, K. Chemotherapy-Induced Constipation and Diarrhea: Pathophysiology, Current and Emerging Treatments. Front. Pharmacol. 2016, 7, 414.
- Van Vliet, M.J.; Tissing, W.J.E.; Dun, C.A.J.; Meessen, N.E.L.; Kamps, W.A.; de Bont, E.S.J.M.; Harmsen, H.J.M. Chemotherapy Treatment in Pediatric Patients with Acute Myeloid Leukemia Receiving Antimicrobial Prophylaxis Leads to a Relative Increase of Colonization with Potentially Pathogenic Bacteria in the Gut. Clin. Infect. Dis. 2009, 49, 262–270.
- Pulito, C.; Cristaudo, A.; Porta, C.L.; Zapperi, S.; Blandino, G.; Morrone, A.; Strano, S. Oral mucositis: The hidden side of cancer therapy. J. Exp. Clin. Cancer Res. 2020, 39, 210.
- Sonis, S.T. The pathobiology of mucositis. Nat. Rev. Cancer 2004, 4, 277–284.
- Al-Asmari, A.K.; Khan, A.Q.; Al-Asmari, S.A.; Al-Rawi, A.; Al-Omani, S. Alleviation of 5-fluorouracil-induced intestinal mucositis in rats by vitamin E via targeting oxidative stress and inflammatory markers. J. Complement. Integr. Med. 2016, 13, 377–385.
- Liu, R.; Bian, Y.; Liu, L.; Liu, L.; Liu, X.; Ma, S. Molecular pathways associated with oxidative stress and their potential applications in radiotherapy. Int. J. Mol. Med. 2022, 49, 65.
- Richardson, R.B.; Harper, M.E. Mitochondrial stress controls the radiosensitivity of the oxygen effect: Implications for radiotherapy. Oncotarget 2016, 7, 21469–21483.
- Musa, A.E.; Shabeeb, D.; Alhilfi, H.S.Q. Protective effect of melatonin against radiotherapy-induced small intestinal oxidative stress: Biochemical evaluation. Medicina 2019, 55, 308.
- Galati, G.; Tafazoli, S.; Sabzevari, O.; Chan, T.S.; O’Brien, P.J. Idiosyncratic NSAID drug induced oxidative stress. Chem.-Biol. Interact. 2002, 142, 25–41.
- Carrasco-Pozo, C.; Gotteland, M.; Speisky, H. Apple peel polyphenol extract protects against indomethacin-induced damage in Caco-2 cells by preventing mitochondrial complex I inhibition. J. Agric. Food Chem. 2011, 59, 11501–11508.
- Nagano, Y.; Matsui, H.; Shimokawa, O.; Hirayama, A.; Tamura, M.; Nakamura, Y.; Kaneko, T.; Rai, K.; Indo, H.; Majima, H. Rebamipide attenuates nonsteroidal anti-inflammatory drugs (NSAID) induced lipid peroxidation by the manganese superoxide dismutase (MnSOD) overexpression in gastrointestinal epithelial cells. J. Physiol. Pharmacol. 2012, 63, 137.
- Klinowski, E.; Broide, E.; Varsano, R.; Eshchar, J.; Scapa, E. Superoxide dismutase activity in duodenal ulcer patients. Eur. J. Gastroenterol. Hepatol. 1996, 8, 1151–1155.
- Uc, A.; Vasiliauskas, E.; Piccoli, D.A.; Flores, A.F.; Di Lorenzo, C.; Hyman, P.E. Chronic intestinal pseudoobstruction associated with fetal alcohol syndrome. Dig. Dis. Sci. 1997, 42, 1163–1167.
- Keshavarzian, A.; Farhadi, A.; Forsyth, C.B.; Rangan, J.; Jakate, S.; Shaikh, M.; Banan, A.; Fields, J.Z. Evidence that chronic alcohol exposure promotes intestinal oxidative stress, intestinal hyperpermeability and endotoxemia prior to development of alcoholic steatohepatitis in rats. J. Hepatol. 2009, 50, 538–547.
- Forsyth, C.B.; Voigt, R.M.; Shaikh, M.; Tang, Y.; Cederbaum, A.I.; Turek, F.W.; Keshavarzian, A. Role for intestinal CYP2E1 in alcohol-induced circadian gene-mediated intestinal hyperpermeability. Am. J. Physiol.-Gastrointest. Liver Physiol. 2013, 305, G185–G195.
- De Vries, D.K.; Kortekaas, K.A.; Tsikas, D.; Wijermars, L.G.; van Noorden, C.J.; Suchy, M.T.; Cobbaert, C.M.; Klautz, R.J.; Schaapherder, A.F.; Lindeman, J.H. Oxidative damage in clinical ischemia/reperfusion injury: A reappraisal. Antioxid. Redox Signal. 2013, 19, 535–545.
- Amaya, Y.; Yamazaki, K.; Sato, M.; Noda, K.; Nishino, T.; Nishino, T. Proteolytic conversion of xanthine dehydrogenase from the NAD-dependent type to the O2-dependent type. Amino acid sequence of rat liver xanthine dehydrogenase and identification of the cleavage sites of the enzyme protein during irreversible conversion by trypsin. J. Biol. Chem. 1990, 265, 14170–14175.
- Sasaki, M.; Joh, T. Oxidative stress and ischemia-reperfusion injury in gastrointestinal tract and antioxidant, protective agents. J. Clin. Biochem. Nutr. 2007, 40, 1–12.
- Chung, H.Y.; Baek, B.S.; Song, S.H.; Kim, M.S.; Huh, J.I.; Shim, K.H.; Kim, K.W.; Lee, K.H. Xanthine dehydrogenase/xanthine oxidase and oxidative stress. Age 1997, 20, 127–140.
- Farias Rolim, M.; Riger, C.J.; Eleutherio, E.C.; da Fonseca Colão, C.; Cotta Pereira, G.; Schanaider, A. Colonic healing after portal ischemia and reperfusion: An experimental study with oxidative stress biomarkers. Redox Rep. 2007, 12, 267–274.
- Bauer, A.J.; Boeckxstaens, G.E. Mechanisms of postoperative ileus. Neurogastroenterol. Motil. 2004, 16 (Suppl. S2), 54–60.
- Kalff, J.C.; Schraut, W.H.; Simmons, R.L.; Bauer, A.J. Surgical manipulation of the gut elicits an intestinal muscularis inflammatory response resulting in postsurgical ileus. Ann. Surg. 1998, 228, 652–663.
- De Backer, O.; Elinck, E.; Blanckaert, B.; Leybaert, L.; Motterlini, R.; Lefebvre, R.A. Water-soluble CO-releasing molecules reduce the development of postoperative ileus via modulation of MAPK/HO-1 signalling and reduction of oxidative stress. Gut 2009, 58, 347.
- Prasad, R.; Kowalczyk, J.; Meimaridou, E.; Storr, H.; Metherell, L. Oxidative stress and adrenocortical insufficiency. J. Endocrinol. 2014, 221, R63.
- Smeitink, J.A.; Zeviani, M.; Turnbull, D.M.; Jacobs, H.T. Mitochondrial medicine: A metabolic perspective on the pathology of oxidative phosphorylation disorders. Cell Metab. 2006, 3, 9–13.
- Welsh, C.; Shifrin, Y.; Pan, J.; Belik, J. Infantile hypertrophic pyloric stenosis (IHPS): A study of its pathophysiology utilizing the newborn hph-1 mouse model of the disease. Am. J. Physiol.-Gastrointest. Liver Physiol. 2014, 307, G1198–G1206.
- Torres, J.; Mehandru, S.; Colombel, J.-F.; Peyrin-Biroulet, L. Crohn’s Disease. Lancet 2017, 389, 1741–1755.
- Ungaro, R.; Mehandru, S.; Allen, P.B.; Peyrin-Biroulet, L.; Colombel, J.-F. Ulcerative Colitis. Lancet 2017, 389, 1756–1770.
- Hancock, L.; Windsor, A.C.; Mortensen, N.J. Inflammatory bowel disease: The view of the surgeon. Color. Dis. 2006, 8, 10–14.
- Lih-Brody, L.; Powell, S.R.; Collier, K.P.; Reddy, G.M.; Cerchia, R.; Kahn, E.; Weissman, G.S.; Katz, S.; Floyd, R.A.; McKinley, M.J.; et al. Increased oxidative stress and decreased antioxidant defenses in mucosa of inflammatory bowel disease. Dig. Dis. Sci. 1996, 41, 2078–2086.
- Piechota-Polanczyk, A.; Fichna, J. Review article: The role of oxidative stress in pathogenesis and treatment of inflammatory bowel diseases. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2014, 387, 605–620.
- Lakhan, S.E.; Kirchgessner, A. Neuroinflammation in inflammatory bowel disease. J. Neuroinflammation 2010, 7, 37.
- Sahakian, L.; Filippone, R.T.; Stavely, R.; Robinson, A.M.; Yan, X.S.; Abalo, R.; Eri, R.; Bornstein, J.C.; Kelley, M.R.; Nurgali, K. Inhibition of APE1/Ref-1 redox signaling alleviates intestinal dysfunction and damage to myenteric neurons in a mouse model of spontaneous chronic colitis. Inflamm. Bowel Dis. 2021, 27, 388–406.
- Koutroubakis, I.E.; Malliaraki, N.; Dimoulios, P.D.; Karmiris, K.; Castanas, E.; Kouroumalis, E.A. Decreased total and corrected antioxidant capacity in patients with inflammatory bowel disease. Dig. Dis. Sci. 2004, 49, 1433–1437.
- Hackam, D.J.; Sodhi, C.P.; Good, M. New insights into necrotizing enterocolitis: From laboratory observation to personalized prevention and treatment. J. Pediatr. Surg. 2019, 54, 398–404.
- Aydemir, C.; Dilli, D.; Uras, N.; Ulu, H.O.; Oguz, S.S.; Erdeve, O.; Dilmen, U. Total oxidant status and oxidative stress are increased in infants with necrotizing enterocolitis. J. Pediatr. Surg. 2011, 46, 2096–2100.
- Perrone, S.; Tataranno, M.L.; Negro, S.; Cornacchione, S.; Longini, M.; Proietti, F.; Soubasi, V.; Benders, M.J.; Van Bel, F.; Buonocore, G. May oxidative stress biomarkers in cord blood predict the occurrence of necrotizing enterocolitis in preterm infants? J. Matern. Fetal Neonatal Med. 2012, 25 (Suppl. S1), 128–131.
- Aceti, A.; Beghetti, I.; Martini, S.; Faldella, G.; Corvaglia, L. Oxidative Stress and Necrotizing Enterocolitis: Pathogenetic Mechanisms, Opportunities for Intervention, and Role of Human Milk. Oxidative Med. Cell. Longev. 2018, 2018, 7397659.
- Silva, K.D.A.; Nunes, J.P.S.; Andrieux, P.; Brochet, P.; Almeida, R.R.; Kuramoto Takara, A.C.K.; Pereira, N.B.; Abel, L.; Cobat, A.; Zaniratto, R.C.F.; et al. Chagas Disease Megaesophagus Patients Carrying Variant MRPS18B P260A Display Nitro-Oxidative Stress and Mitochondrial Dysfunction in Response to IFN-γ; Stimulus. Biomedicines 2022, 10, 2215.
- Weimers, P.; Munkholm, P. The Natural History of IBD: Lessons Learned. Curr. Treat. Options Gastroenterol. 2018, 16, 101–111.
- Yalchin, M.; Baker, A.M.; Graham, T.A.; Hart, A. Predicting Colorectal Cancer Occurrence in IBD. Cancers 2021, 13, 2908.
- Carini, F.; Mazzola, M.; Rappa, F.; Jurjus, A.; Geagea, A.G.; Al Kattar, S.; Bou-Assi, T.; Jurjus, R.; Damiani, P.; Leone, A. Colorectal carcinogenesis: Role of oxidative stress and antioxidants. Anticancer Res. 2017, 37, 4759–4766.
- Peng, D.; Zaika, A.; Que, J.; El-Rifai, W. The antioxidant response in Barrett’s tumorigenesis: A double-edged sword. Redox Biol. 2021, 41, 101894.
- Jiménez, P.; Piazuelo, E.; Sánchez, M.T.; Ortego, J.; Soteras, F.; Lanas, A. Free radicals and antioxidant systems in reflux esophagitis and Barrett’s esophagus. World J. Gastroenterol. 2005, 11, 2697–2703.
- Skrzydlewska, E.; Sulkowski, S.; Koda, M.; Zalewski, B.; Kanczuga-Koda, L.; Sulkowska, M. Lipid peroxidation and antioxidant status in colorectal cancer. World J. Gastroenterol. 2005, 11, 403–406.
- Bhor, V.M.; Raghuram, N.; Sivakami, S. Oxidative damage and altered antioxidant enzyme activities in the small intestine of streptozotocin-induced diabetic rats. Int. J. Biochem. Cell Biol. 2004, 36, 89–97.
- Stavely, R.; Abalo, R.; Nurgali, K. Targeting Enteric Neurons and Plexitis for the Management of Inflammatory Bowel Disease. Curr. Drug Targets 2020, 21, 1428–1439.
- Furness, J.B. The Enteric Nervous System; Blackwell Publishing: Hoboken, NJ, USA, 2006.
- Schneider, S.; Wright, C.M.; Heuckeroth, R.O. Unexpected Roles for the Second Brain: Enteric Nervous System as Master Regulator of Bowel Function. Annu. Rev. Physiol. 2019, 81, 1–25.
- Goldstein, A.M.; Thapar, N.; Karunaratne, T.B.; De Giorgio, R. Clinical aspects of neurointestinal disease: Pathophysiology, diagnosis, and treatment. Dev. Biol. 2016, 417, 217–228.
- Pellegrini, C.; D’Antongiovanni, V.; Miraglia, F.; Rota, L.; Benvenuti, L.; Di Salvo, C.; Testa, G.; Capsoni, S.; Carta, G.; Antonioli, L.; et al. Enteric α-synuclein impairs intestinal epithelial barrier through caspase-1-inflammasome signaling in Parkinson’s disease before brain pathology. NPJ Park. Dis. 2022, 8, 9.
- Rota, L.; Pellegrini, C.; Benvenuti, L.; Antonioli, L.; Fornai, M.; Blandizzi, C.; Cattaneo, A.; Colla, E. Constipation, deficit in colon contractions and alpha-synuclein inclusions within the colon precede motor abnormalities and neurodegeneration in the central nervous system in a mouse model of alpha-synucleinopathy. Transl. Neurodegener. 2019, 8, 5.
- Friedman, J. Why is the nervous system vulnerable to oxidative stress. In Oxidative Stress and Free Radical Damage in Neurology; Springer: Berlin/Heidelberg, Germany, 2011; pp. 19–27.
- Brown, I.A.M.; McClain, J.L.; Watson, R.E.; Patel, B.A.; Gulbransen, B.D. Enteric Glia Mediate Neuron Death in Colitis Through Purinergic Pathways That Require Connexin-43 and Nitric Oxide. Cell. Mol. Gastroenterol. Hepatol. 2016, 2, 77–91.
- Wada-Takahashi, S.; Tamura, K. Actions of reactive oxygen species on AH/type 2 myenteric neurons in guinea pig distal colon. Am. J. Physiol.-Gastrointest. Liver Physiol. 2000, 279, G893–G902.
- Roberts, J.A.; Durnin, L.; Sharkey, K.A.; Mutafova-Yambolieva, V.N.; Mawe, G.M. Oxidative stress disrupts purinergic neuromuscular transmission in the inflamed colon. J. Physiol. 2013, 591, 3725–3737.
- Thrasivoulou, C.; Soubeyre, V.; Ridha, H.; Giuliani, D.; Giaroni, C.; Michael, G.J.; Saffrey, M.J.; Cowen, T. Reactive oxygen species, dietary restriction and neurotrophic factors in age-related loss of myenteric neurons. Aging Cell 2006, 5, 247–257.
- Ricci, M.F.; Béla, S.R.; Moraes, M.M.; Bahia, M.T.; Mazzeti, A.L.; Oliveira, A.C.S.; Andrade, L.O.; Radí, R.; Piacenza, L.; Arantes, R.M.E. Neuronal Parasitism, Early Myenteric Neurons Depopulation and Continuous Axonal Networking Damage as Underlying Mechanisms of the Experimental Intestinal Chagas’ Disease. Front. Cell. Infect. Microbiol. 2020, 10, 583899.
- Chandrasekharan, B.; Anitha, M.; Blatt, R.; Shahnavaz, N.; Kooby, D.; Staley, C.; Mwangi, S.; Jones, D.P.; Sitaraman, S.V.; Srinivasan, S. Colonic motor dysfunction in human diabetes is associated with enteric neuronal loss and increased oxidative stress. Neurogastroenterol. Motil. 2011, 23, 131-e26.
- McQuade, R.M.; Stojanovska, V.; Stavely, R.; Timpani, C.; Petersen, A.C.; Abalo, R.; Bornstein, J.C.; Rybalka, E.; Nurgali, K. Oxaliplatin-induced enteric neuronal loss and intestinal dysfunction is prevented by co-treatment with BGP-15. Br. J. Pharmacol. 2018, 175, 656–677.
- Linden, D.; Couvrette, J.; Ciolino, A.; McQuoid, C.; Blaszyk, H.; Sharkey, K.; Mawe, G. Indiscriminate loss of myenteric neurones in the TNBS-inflamed guinea-pig distal colon. Neurogastroenterol. Motil. 2005, 17, 751–760.
- Winston, J.H.; Li, Q.; Sarna, S.K. Paradoxical regulation of ChAT and nNOS expression in animal models of Crohn’s colitis and ulcerative colitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 305, G295–G302.
- Stavely, R.; Robinson, A.M.; Miller, S.; Boyd, R.; Sakkal, S.; Nurgali, K. Human adult stem cells derived from adipose tissue and bone marrow attenuate enteric neuropathy in the guinea-pig model of acute colitis. Stem Cell Res. Ther. 2015, 6, 1–21.
- Stavely, R.; Robinson, A.M.; Miller, S.; Boyd, R.; Sakkal, S.; Nurgali, K. Allogeneic guinea pig mesenchymal stem cells ameliorate neurological changes in experimental colitis. Stem Cell Res. Ther. 2015, 6, 263.
- Bubenheimer, R.K.; Brown, I.A.M.; Fried, D.E.; McClain, J.L.; Gulbransen, B.D. Sirtuin-3 Is Expressed by Enteric Neurons but It Does not Play a Major Role in Their Regulation of Oxidative Stress. Front. Cell. Neurosci. 2016, 10, 73.
- Nurgali, K.; Qu, Z.; Hunne, B.; Thacker, M.; Pontell, L.; Furness, J.B. Morphological and functional changes in guinea-pig neurons projecting to the ileal mucosa at early stages after inflammatory damage. J. Physiol. 2011, 589, 325–339.
- Qu, Z.-D.; Thacker, M.; Castelucci, P.; Bagyánszki, M.; Epstein, M.L.; Furness, J.B. Immunohistochemical analysis of neuron types in the mouse small intestine. Cell Tissue Res. 2008, 334, 147–161.
- Wafai, L.; Taher, M.; Jovanovska, V.; Bornstein, J.C.; Dass, C.R.; Nurgali, K. Effects of oxaliplatin on mouse myenteric neurons and colonic motility. Front. Neurosci. 2013, 7, 30.
- Rivera, L.R.; Poole, D.P.; Thacker, M.; Furness, J.B. The involvement of nitric oxide synthase neurons in enteric neuropathies. Neurogastroenterol. Motil. 2011, 23, 980–988.
- Rivera, L.R.; Thacker, M.; Pontell, L.; Cho, H.-J.; Furness, J.B. Deleterious effects of intestinal ischemia/reperfusion injury in the mouse enteric nervous system are associated with protein nitrosylation. Cell Tissue Res. 2011, 344, 111–123.
- Bagyánszki, M.; Krecsmarik, M.; De Winter, B.Y.; De Man, J.G.; Fekete, E.; Pelckmans, P.A.; Adriaensen, D.; Kroese, A.B.; Van Nassauw, L.; Timmermans, J.P. Chronic alcohol consumption affects gastrointestinal motility and reduces the proportion of neuronal NOS-immunoreactive myenteric neurons in the murine jejunum. Anat. Record 2010, 293, 1536–1542.
- Izbéki, F.; Wittman, T.; Rosztóczy, A.; Linke, N.; Bódi, N.; Fekete, E.; Bagyánszki, M. Immediate insulin treatment prevents gut motility alterations and loss of nitrergic neurons in the ileum and colon of rats with streptozotocin-induced diabetes. Diabetes Res. Clin. Pract. 2008, 80, 192–198.
- Bódi, N.; Szalai, Z.; Bagyánszki, M. Nitrergic Enteric Neurons in Health and Disease-Focus on Animal Models. Int. J. Mol. Sci. 2019, 20, 2003.
- Berghe, P.V.; Kenyon, J.L.; Smith, T.K. Mitochondrial Ca2+ uptake regulates the excitability of myenteric neurons. J. Neurosci. 2002, 22, 6962–6971.
- Aon, M.A.; Cortassa, S.; O’Rourke, B. Redox-optimized ROS balance: A unifying hypothesis. Biochim. Biophys. Acta 2010, 1797, 865–877.
- Jia, J.; Jin, H.; Nan, D.; Yu, W.; Huang, Y. New insights into targeting mitochondria in ischemic injury. Apoptosis 2021, 26, 163–183.
- Kwak, D.J.; Kwak, S.D.; Gauda, E.B. The effect of hyperoxia on reactive oxygen species (ROS) in rat petrosal ganglion neurons during development using organotypic slices. Pediatr. Res. 2006, 60, 371–376.
- D’Agostino, D.P.; Putnam, R.W.; Dean, J.B. Superoxide (O2.−) production in CA1 neurons of rat hippocampal slices exposed to graded levels of oxygen. J. Neurophysiol. 2007, 98, 1030–1041.
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13.
- Matott, M.P.; Ciarlone, G.E.; Putnam, R.W.; Dean, J.B. Normobaric hyperoxia (95% O2) stimulates CO2-sensitive and CO2-insensitive neurons in the caudal solitary complex of rat medullary tissue slices maintained in 40% O2. Neuroscience 2014, 270, 98–122.
- Chang, E.; Hornick, K.; Fritz, K.I.; Mishra, O.P.; Delivoria-Papadopoulos, M. Effect of hyperoxia on cortical neuronal nuclear function and programmed cell death mechanisms. Neurochem. Res. 2007, 32, 1142–1149.
- Sundaram, U.; Hassanain, H.; Suntres, Z.; Yu, J.G.; Cooke, H.J.; Guzman, J.; Christofi, F.L. Rabbit chronic ileitis leads to up-regulation of adenosine A1/A3 gene products, oxidative stress, and immune modulation. Biochem. Pharmacol. 2003, 65, 1529–1538.