Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Halina Leung and Version 2 by Fanny Huang.
Neutrophil extracellular traps (NETs) are major contributors to inflammation and autoimmunity, playing a key role in the development of thrombotic disorders. NETs, composed of DNA, histones, and numerous other proteins serve as scaffolds for thrombus formation and promote platelet activation, coagulation, and endothelial dysfunction.
- immune complex
- neutrophil extracellular traps
- Fc receptors
- thrombosis
1. Introduction
Morphological changes in neutrophils, distinct from apoptosis or necrosis, upon treatment with phorbol 12-myristate 13-acetate (PMA) were described by Takei et al. in 1996 [1]. This process was later termed neutrophil extracellular traps (NETs) formation and was initially reported as a host defence mechanism against infection [2]. NETs formation has evolved into a mechanism with wide implications in biological processes and in the pathology of diabetes, cancer, autoimmunity, atherosclerosis, infections, and thrombosis. Two distinct types of NETosis have been described: a slow form of cell death that may take hours and is known as lytic or suicidal NETosis and vital NETosis, a fast process whereby neutrophils release nuclear material but remain viable and functional [3]. In fact, enucleated neutrophils are known to retain chemotactic and phagocytic functions [4].
Lytic NETosis depends on NADPH and reactive oxygen species (ROS) activity which triggers histone citrullination via peptidyl arginine deiminase (PAD4), while vital NETosis does not involve NADPH and relies on increases in intracellular calcium [5]. A sub-category of lytic NETosis, termed noncanonical NETosis, has been proposed more recently [6]. This form of NETosis is elicited in the presence of cytosolic Gram-negative bacteria and involves noncanonical inflammasomes and caspase 4/5 activation. Histone citrullination mediated by PAD4 is also present in noncanonical NETosis [7]. Both lytic and vital NETosis, however, involve nuclear the decondensation and subsequent release of chromatin into the extracellular space. Chromatin serves as the backbone for numerous NET components including histones, myeloperoxidase, elastase, and cathepsin-G [8]. Therefore, the consequences of NETs formation—antibacterial activity, inflammation, vascular occlusion—can be attributed to the activity of the diverse components of these DNA structures.
Since the description of NETs induction by PMA and bacteria [1][2][1,2], numerous stimuli of NETs formation such as viruses [9], cholesterol crystals [10], activated platelets [11][12][11,12], cytokines [13], autoantibodies [14], and immune complexes (ICs) [15][16][17][15,16,17] have been described. Unsurprisingly, an attendant number of neutrophil receptors have been associated with various stimuli. Activated platelets induce NETosis via the PSGL1 receptor [15][18][15,18] or via high-mobility group protein B1-RAGE interaction [19], bacteria and viruses through pattern recognition receptors and Fc receptors [20][21][20,21] and ICs via Fc receptors [15][16][22][15,16,22] (Figure 1).
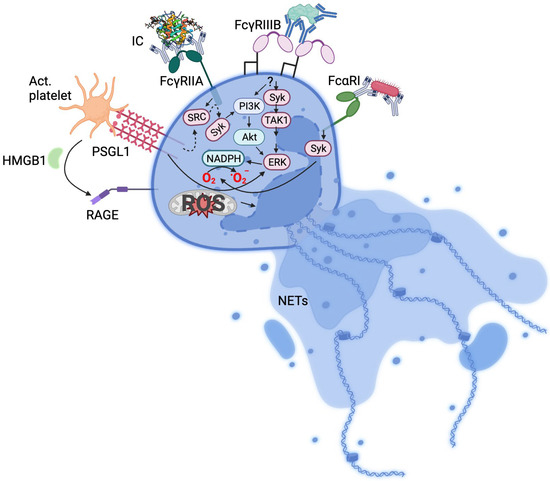
Figure 1. Mechanisms of neutrophil activation. Summarised receptor signalling pathways implicated in NETosis. IC, immune complex; Act. platelet, activated platelets; HMB1, high-mobility group box 1; TAK1, transforming growth factor-β-activated kinase 1; RAGE, receptor for advanced glycation end-products; PSGL-1, P-selectin glycoprotein ligand-1; dotted arrows indicate intermediate signalling molecules not included in the Figure.
2. Immune Complexes and Fc Receptors
The adaptive immune response results in the production of antibodies against antigens, leading to the formation of antigen–antibody complexes. The generation of antibodies that bind to different epitopes on the antigen leads to the formation of ICs. The role of ICs in conditions such a serum sickness, vasculitis, and rheumatic disease has been documented for decades [23]. ICs have a higher affinity for Fc receptors and are normally cleared by the liver (by sinusoidal endothelial cells and Kupffer cells) and spleen via interaction with Fc receptors expressed on monocytes/macrophages and neutrophils. Neutrophils express both low- (FcγRIIA, FcγRIIB, and FcγRIIIB) and high- affinity (FcRn and FcγRI, which is expressed on activated neutrophils) IgG Fc receptors. FcαRI, a receptor for IgA, is also present on human neutrophils [24]. High-affinity receptors such as FcγRI bind IgG monomers, while the low affinity receptors have an avidity for IgG ICs or opsonised cells. IgG subclasses display distinct Fc receptor binding affinities. IgG1 and IgG3 interact with FcγRI, FcγRIIA/B, FcγRIIIB, and FcRn, while IgG2 fails to recognise FcγRI, FcγRIIB, and FcγRIIIB [25]. Some IgG2 and IgG4 ICs also recognise complement receptors on neutrophils [26].
The recognition of ICs by Fc receptors leads to cellular signalling. For example, the clustering of FcγRIIA via interaction with ICs induces a phosphorylation of the immunoreceptor tyrosine-based activation motif (ITAM) by Src kinases [27], leading to Syk signalling involving PI3K and PLCγ. This triggers diverse responses including phagocytosis and receptor internalisation, cytokine production, and oxidative burst. Murine neutrophils expressing human FcγRIIA and FcγRIIIB are able to uptake ICs, but only FcγRIIA and not FcγRIIIB transgenic mice formed NETs in response to ICs in vivo [28]. In human neutrophils, however, FcγRIIIB has been found to be involved in NETs formation [29].
Circulating ICs resulting from excessive antibody production or clearance failure can be deposited on tissues, causing inflammation, and as such are involved in the pathology of multiple conditions including autoimmune diseases such as arthritis and lupus erythematosus. Drug-mediated reactions such as heparin-induced thrombocytopenia (HIT) and vaccine-induced thrombotic thrombocytopenia (VITT) and infections including influenza and SARS-CoV-2 are facilitated by circulating ICs. In fact, the induction of platelet activation via IC activity has been known since the 1950s [30].
It should be noted that ICs also induce processes analogous to NETosis in monocytes [31]. Monocyte extracellular traps (ETs) possess a procoagulant activity, suggesting a role for these monocyte-derived structures in thrombosis (reviewed by Han et al. [32]). The more generalised nature of ET formation and its association with thrombosis is exemplified by observations of ETs of macrophage, mast cell, and eosinophil origin in coronary thrombi [33].
IgG, and more specifically IgG1, is the most common type of immunoglobulin in human serum [25]. IgG ICs have been more widely described, and it is reasonable to assume that IgG ICs are also the most abundant. Lupus erythematosus is an extensively studied autoimmune disease characterised by the presence of autoantibodies against numerous endogenous antigens including dsDNA, ribonucleoprotein, phospholipids, histones, and β2-glycoprotein I. Most of these autoantibodies are of the IgG class [34]. DNA-containing Ics found in lupus patients signal through FcγRIIA and induce ROS production in neutrophils [35]. This is reminiscent of ROS-dependent neutrophil activation, NETosis, and thrombosis induced by FcγRIIA-activating HIT Ics [36].
ADAMTS-13 ICs in thrombotic thrombocytopenic purpura are correlated with relapse [37] and markers of NETosis such as histone/DNA complexes, cell-free DNA, and citrullinated histones are present in plasma from these patients [38], suggesting a contribution of ICs in the inductions of NETosis and thrombosis in this condition. IgG ICs are also present in granulomatosis (antineutrophil cytoplasmic antibodies [39]), SARS-CoV-2 infection [40], rheumatoid arthritis [41], HIT, and VITT [15][16][15,16] (Table 1).
Thrombosis is also driven by IgA ICs (Table 1). Antibody specificity and signalling is dependent on the isotype, where IgG binds to FcγR while IgA binds to FcαR. In the context of IgG signalling, unlike low-affinity FcγRs, only high-affinity FcγRs (i.e., FcγR1) can bind monomeric IgG. All FcγRs can, however, bind to IgG aggregates or immune complexes containing IgG [42]. Similarly, monomeric IgA binds poorly to FcαR1, while large IgA complexes bind with a high avidity, leading to phagocytosis, antigen presentation [43], cytokine release [44], reactive oxygen species production [45], and NETosis [46][47][48][46,47,48].
The proinflammatory IgA immune complex-mediated FcαR signalling is a key pathogenic feature in IgA vasculitis (or Henoch–Schönlein purpura), an inflammatory condition where the immune system attacks the lining of blood vessels. Recently, Mayer-Hain et al. showed the requirement of neutrophil prestimulation by polymeric IgA or IgA ICs to lower the threshold for neutrophil activation. This step is critical for neutrophils to become activated and undergo NETosis upon binding to activated endothelial cells, resulting in vessel wall damage [46]. Interestingly, neutrophils isolated from IgA vasculitis patients spontaneously underwent NETosis, and NETs were proximal to endothelial cells and IgA-coated neutrophils in tissue sections of these patients [46]. A significant reduction in vessel damage in a mouse model of vasculitis was observed following NET inhibition, suggesting that NETosis is key a mediator of vasculitis pathogenesis. Although uncommon, cases of coagulation abnormalities and thrombosis have been documented in IgA vasculitis [49]. Altogether, IgA ICs prestimulate neutrophils and NETosis is a key mediator of vessel wall damage in IgA vasculitis.
The presence of IgA autoantibodies is also associated with increased disease severity, enhanced cartilage damage, and worse disease prognosis in rheumatoid arthritis [50][51][52][50,51,52]. The activation of neutrophils by IgA ICs, present in rheumatoid arthritis patients’ plasma and synovial fluid, leads them to undergo NETosis and secrete chemoattractants that amplify neutrophil recruitment [47] and promotes cartilage damage via neutrophil elastase [48]. IgA IC-induced NETosis can be blocked by anti-FcαR1 monoclonal antibody [47], suggesting FcαR1 inhibitors could potentially reduce cartilage damage and disability in rheumatoid arthritis patients. In antiphospholipid syndrome (APS), β2-glycoprotein I/IgA IC is strongly linked to thrombosis following transplantation [53]. NETosis is likely to be of pathological significance since NETs are known to contribute to thrombosis in APS [54].
NETosis has been well documented in bacterial (e.g., Staphylococcus aureus [55]) and viral (e.g., influenza A, HIV, SARS-CoV-2 [21]) infections. NETs exert both antimicrobial [56] and antiviral [21] activity. IgA-bacteria ICs activate FcαRI and enhance the phagocytosis of IgA-opsonized bacteria [57]. Interestingly, the phagocytosis of S. aureus is more efficient in the presence of IgA [58]. Both bacterial and viral IgA IC-induced NETosis are dependent on FcαRI and NADPH oxidase [21][58][21,58]. A lower virus titre is required to trigger NETosis in the presence of IgA compared to virus alone, and NETosis is independent of phagocytosis. Unlike NETosis induced by virus alone, IgA-virus ICs-induced NETosis does not require toll-like receptor signalling [21]. Although NETosis plays a protective role in infection, the release of reactive oxygen species, proteolytic enzymes and inflammatory mediators can enhance neutrophil infiltration and elicit tissue damage. This can result in the enhancement of disease pathogenesis in conditions such as respiratory syncytial virus [59], rhinovirus [60], influenza [61], and COVID-19 [62][63][64][62,63,64].
Table 1. Fc receptors implicated in IgG- and IgA-immune complex-mediated NETosis.
| Condition | Fc Receptor Involved | Receptor Binding Specificity | Antigen-Antibody Complex | Reference |
|---|---|---|---|---|
| Influenza A | FcαR1 (CD89) | IgA | Influenza—IgA | Stacey et al. [21] |
| Human immunodeficiency virus (HIV) | FcαR1 (CD89) | IgA | HIV—IgA | Stacey et al. [21] |
| SARS-CoV-2 | FcαR1 (CD89) FcγRIIA (CD32a) |
IgA IgG |
SARS-CoV2—IgA Spike protein—IgG |
Stacey et al. [21] Bye et al. [65] |
| Staphylococcus aureus | FcαR1 (CD89) | IgA | S. aureus—IgA | Aleyd et al. [58] |
| Vasculitis | FcαR1 (CD89) | IgA | Aggregated vasculitis—IgA | Mayer-Hain et al. [46] |
| Rheumatoid arthritis | FcαR1 (CD89), FcγRI (CD64), FcγRIIA (CD32a) | IgA, IgG | Rheumatoid factor—IgA/IgG, citrullinated protein—anticitrullinated protein antibody (ACPA) IgG, cyclic citrullinated peptide—IgA/IgG, antineutrophil cytoplasmic antibodies (ANCA)—IgA/IgG | Mathsson et al. [41]; Aleyd et al. [47]; Kempers et al. [66] |
| Granulomatosis with polyangiitis (Wegener’s granulomatosis) | FcαR1 (CD89), FcγRIIIB (CD16b) | IgA, IgG | Antineutrophil cytoplasmic antibodies (ANCA)—IgA/IgG | Kelley et al. [39] |
| Heparin-induced thrombocytopenia (HIT) | FcγRIIA (CD32a) | IgG | Heparin—PF4 -HIT IgG | Kelton et al. [67]; Chong et al. [68] |
| Vaccine-induced thrombotic thrombocytopenia (VITT) | FcγRIIA (CD32a) | IgG | PF4—VITT IgG | Greinacher et al. [69] |
| Autoimmune inflammatory disorder | FcγRIIA (CD32a), FcγRIIIB (CD16b) | IgG | Bovine serum albumin (BSA)—IgG, human serum albumin (HAS)—IgG, cross linking—FcγRIIIB | Aleman et al. [29]; Behnen et al. [22] |
| Systemic lupus erythematosus (SLE) | FcγRIIA (CD32a) | IgG | DNA—IgG | Bonegio et al. [35]; Bruneau et al. [70]; Patiño-Trives et al. [71]; Dema and Charles [34]. |