Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Javier Ramirez Jirano and Version 3 by Alfred Zheng.
Frontotemporal lobar degeneration (FTLD) belongs to a heterogeneous group of highly complex neurodegenerative diseases and represents the second cause of presenile dementia in individuals under 65. Frontotemporal-TDP is a subgroup of frontotemporal dementia characterized by the aggregation of abnormal protein deposits, predominantly transactive response DNA-binding protein 43 (TDP-43), in the frontal and temporal brain regions. These deposits lead to progressive degeneration of neurons resulting in cognitive and behavioral impairments. Limbic age-related encephalopathy (LATE) pertains to age-related cognitive decline primarily affecting the limbic system, which is crucial for memory, emotions, and learning.
- frontotemporal lobar degeneration
- transactive response DNA-binding protein 43
1. Biology of TDP-43
1.1. Structure of TDP-43
Transactive response DNA-binding protein 43 (TDP-43) is a highly conserved, predominantly nuclear protein encoded by the TAR-DNA-binding protein (TARDBP) gene found on chromosome 1. The TDP-43 name comes from its discovery when it was identified as a human trans-activation response element (TAR) binding protein [1]. TDP-43 belongs to the family of heterogeneous nuclear ribonucleoproteins (hnRNPs), which are proteins that primarily interact with pre-messenger transcripts. The TARDBP gene encodes a single isoform of 414 amino acids. Structurally, TDP-43 has two ribonucleic acid (RNA)-binding domains (RRM1 and RRM2, where RRM stands for RNA recognition motif) and a glycine-rich C-terminal region used to interact with other proteins such as hnRNP A1/A2 and C1. This region also contains a prion-like domain (rich in glutamine/asparagine) that plays an important role in TDP-43 aggregation [2]. Also, TDP-43 has nuclear export (NES) and nuclear localization (NLS) sequences necessary for its passage from the nucleus to the cytoplasm and vice versa.
1.2. Physiological Functions of TDP-43
Described initially as a DNA-binding protein, TDP-43 is now known to regulate nucleic acid metabolism through different mechanisms, primarily:
1.2.1. Nuclear Functions: Modulation of Pre-mRNA Splicing by TDP-43
In 2001, a study highlighted the participation of TDP-43 in splicing exon 9 of the pre-mRNA encoded by the cystic fibrosis transmembrane conductance regulator (CFTR) gene [3]. Subsequently, the use of the high-throughput sequencing of RNA isolated by the crosslinking immunoprecipitation (HITS-CLIP) technique allows the identification of RNA bound to a protein, revealing that TDP-43 interacts with a large part of the nervous system transcriptome (central nervous system (CNS)) (>6000 RNA). Interestingly, most of these RNAs involve essential cellular processes such as neuronal development, axonal guidance, and synaptic activity [4]. Furthermore, many of these transcripts show splicing changes in FTLD with amyotrophic lateral sclerosis (ALS) patients and TDP-43-deficient mouse models [5][6][5,6]. TDP-43 binds mainly at the level of introns and 3′UTR regions (untranslated regions) on sequences preferably rich in UG repeats, although this feature is not essential for interaction. Thus, TDP-43 can interact with its own mRNA’s 3′UTR region to modulate its translation capacity. This autoregulatory mechanism is essential in maintaining the amount of TDP-43 in the cell at a physiological level [7].
1.2.2. Nuclear Functions: Regulation of Micro-RNAs
Less than 5% of the RNA produced inside cells codes for proteins. It is now known that the remaining 95%, called non-coding RNAs (ncRNAs), play an essential role in regulating gene expression within the CNS, both under physiological and pathological conditions [8]. Micro-RNAs (miRNAs) are part of these ncRNAs, and their function is to repress the expression of the target mRNAs by pairing directly with the complementary sequences to them [9]. The synthesis of miRNAs involves several steps between the nucleus and the cytoplasm and mainly involves two RNase IIIs called Drosha and Dicer. In fact, after the transcription of the primary miRNA (pri-miRNA), the latter is cleaved by the Drosha protein and forms a sequence called pre-miRNA [10]. This pre-miRNA is then transported to the cytoplasm where Dicer will mature it, resulting in a functional 22 nucleotide miRNA [11].
The hypothesis that TDP-43 could participate in the regulation of miRNAs was raised by its presence within the Drosha complex and its association at the level of perichromatin fibrils, where pri-miRNAs are born [12]. In agreement with this first observation, studies have shown deregulation of the expression of various miRNAs, such as let-7b, miR-663, or miR-132 [12]. Most of these deregulations result from a direct interaction between TDP-43 with miRNAs or their precursors. However, during in vitro neuronal differentiation, TDP-43 was shown to modify Drosha stability, suggesting a potential impact on all; in addition, increased Dicer expression was also observed upon TDP-43 repression in a human neuronal cell line model [13]. Together, these data support the critical role of TDP-43 in miRNA metabolism.
1.2.3. Nuclear Functions: Regulation of the Expression of Long Non-Coding RNAs
Among the other ncRNAs, long non-coding RNAs (lncRNAs) are involved in numerous cellular mechanisms, particularly at the transcriptional and post-transcriptional levels. Recently, the deregulation of these lncRNAs has been demonstrated, particularly in pathologies related to aging, including neurodegenerative diseases [14]. In this context, TDP-43 has been shown to affect the expression of several lncRNAs such as MALAT1 (associated with SC35 domains called speckles) and NEAT1_2 (involved in the formation of paraspeckles) [6]. In fact, in the brain of FTLD/ALS patients, TDP-43 binds more to these lncRNAs, and increased expression of NEAT 1_2 was observed in motor neurons of ALS patients, compared to healthy individuals [15]. Given the importance of specks and parameters in RNA metabolism, dysregulation of these lncRNAs could contribute to the pathophysiology of FTLD/ALS [16]. However, although the direct interaction of TDP-43 with these lncRNAs has been established, the mechanisms underlying their dysregulation and how this contributes to neurodegeneration remain to be determined [17]. Although TDP-43 is primarily nuclear, it is also credited with several functions in the cytoplasm including regulation of mRNA stability, transport, and translation.
1.2.4. Cytosolic Functions: Stabilization of mRNA
Transactive response DNA-binding protein 43 was first identified as a regulator of the stability of the mRNA encoding neurofilament lumen (NF-L) and histone deacetylase HDAC6 [18]. Studies demonstrated that 3′UTR regions of mRNAs, which play a crucial role in mRNA stability mediated by RNA-binding proteins (RBPs), were a preferential target of TDP-43 [4][13][4,13]. As a result, other targets of TDP-43, such as β-adducin (Add2) [19], vascular endothelial growth factor A (VEGFA), GRN [20], or even interleukin-6 (IL-6) [21], were discovered.
1.2.5. Cytosolic Functions: Transport of mRNA
Within polarized cells such as neurons, transporting mRNAs to axons and dendrites is a fundamental process in maintaining neuronal activity and synaptic plasticity [22]. Evidence suggests that TDP-43 is involved in this process. First, a study on cultures of primary motor neurons demonstrated the transport of TDP-43 along axons. Interestingly, this study also showed that TDP-43 co-localized with the survival motor neuron protein (SMN) and fragile-X mental retardation protein (FMRP), both of which are known to play a role in mRNA transport within specific structures called mRNA granules [23]. Transport of positive TDP-43 mRNA granules along axons in motor neurons derived from human inducible pluripotent stem cells (iPSCs) was then confirmed. Among the mRNAs present within these granules, the NF-L mRNA, whose stability is regulated by TDP-43, stood out significantly.
Furthermore, the presence of TDP-43 mutations within these iPSCs induces impaired anterograde granule transport, indicating a direct role for TDP-43 in axonal mRNA transport [24]. TDP-43 was also bound to mRNAs encoding proteins related to synaptic function in adult mouse brains. These data, associated with the localization of TDP-43 at the level of the axonal terminals, therefore indicate that TDP-43 plays a role in the transport of RNAs toward the extensions of neurites (Figure 1).
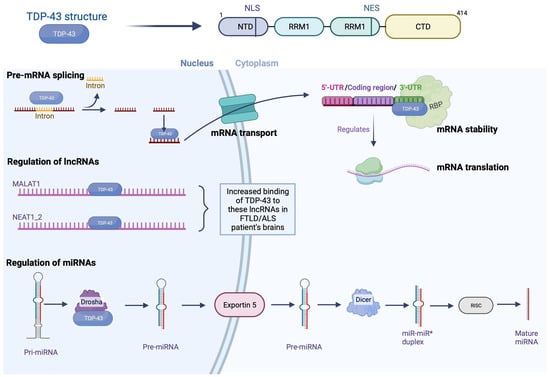
Figure 1. Physiological functions of TDP-43. TPD-43 is a nuclear protein with an N-terminal domain (NTD), two RNA recognition motifs (RRM1 and RRM2), and a long C-terminal domain (CTD). TDP-43 regulates the metabolism of nucleic acid by different mechanisms such as pre-miRNA splicing (TDP-43 binds to introns and 3′UTR regions); it binds to mRNA formed and transports it from the nucleus to the cytoplasm through pores. In the cytoplasm, it gives stability to mRNA by binding to 3′UTR region and is believed to regulate the mRNA translation. In addition, TDP-43 has been shown to bind to long non-coding RNAs such as MALAT1 and NEAT1_2 in neurodegenerative diseases; binding is increased in the brain of FTLD/ALS patients. Finally, it helps in the regulation of miRNAs; the primary miRNA (pri-miRNA) is cleaved by Drosha, and the pre-miRNA is formed, which is transported to the cytoplasm by Exportin 5, where it will mature by Dicer, followed by RISC, and result in a functional miRNA of 22 nucleotides. Nuclear export sequence (NES); nuclear localization sequence (NLS); RNA-binding protein (RBP). Created with Biorender.com.
1.2.6. Cytosolic Functions: Regulation of mRNA Translation
One of the emerging functions of TDP-43, directly related to its role in mRNA transport to distal regions, is the regulation of local mRNA translation. The first proof of their involvement was highlighted in 2008 when a team observed a TDP-43 translocation at the dendrite level after potassium chloride stimulation within rat hippocampal neurons [25]. Furthermore, this study indicated that TDP-43 could repress mRNA translation within mRNA granules. Proteomic analyzes confirmed these results by revealing the interaction between TDP-43 and the translation machinery [26]. A study using cell cultures subjected to oxidative stress also demonstrated the presence of TDP-43 within inactive ribosome-enriched polysomes [27]. More recently, the use of an ALS model in Drosophila has described a regulation of the translation of the Futsch/22C10 microtubule-associated proteins 1B (MAP1B) and Rac1 proteins by TDP-43 [28][29][28,29]. These proteins are involved in neuronal functioning, modulating the formation of dendrites and the architecture of neuromuscular junctions. Therefore, TDP-43 dysregulation could play a crucial role in the pathophysiology of FTLD. A study of the translational profile in murine models of ALS also indicates the deregulation of two mRNAs bound by TDP-43 at the motor neuron level, encoding the DDX58 and MTHFSD proteins, respectively [30] (Figure 1).
1.3. Pathophysiology of TDP-43
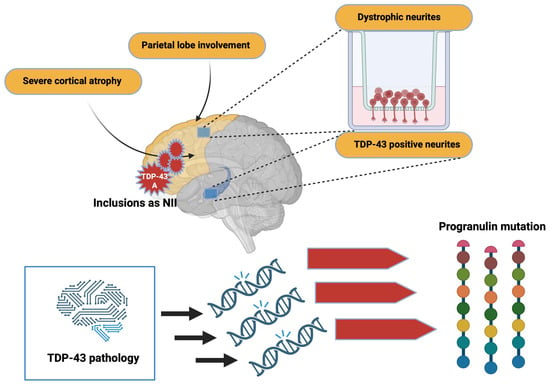
Aggregated forms of TDP-43 exhibit several abnormal changes including hyperphosphorylation, ubiquitination, and N-terminal proteolysis [31]. At the histopathological level, the lesions observed are mainly of the cytoplasmic (NCIs) and neuritic (due to dystrophic neurites (DN)) type, and show immunoreactivity for both TDP-43 and p62 (a protein linked to autophagy). In some cases, usually hereditary, neuronal intranuclear inclusions (NII) are also present. Moreover, other types of lesion that are not ubiquitin-positive have also been demonstrated. Among them can be distinguished diffuse cytoplasmic markings (called pre-inclusions), neuritic markings, and cytoplasmic inclusions within oligodendrocytes (glial cytoplasmic inclusion (GCI)) [32].
Regarding the distribution of lesions, these are commonly found in the frontal and temporal cortex and in the dentate gyrus of the hippocampus. However, many other subcortical regions may also be affected [33]. Depending on the distribution of the inclusions within the cortical layers, and the nature of the inclusions, four histopathological subtypes are defined; each of these subtypes may be partially correlated with a specific clinical syndrome or genetic mutation [32].
2. The Different Histopathological Subtypes of FTLD-TDP
2.1. FTLD-TDP Type A
Type-A FTLD-TDP is distinguished by the presence of numerous DNs and cytoplasmatic aggregates at the level of the second layer of the neocortex. A moderate number of NCI granular inclusions are also present within the dentate gyrus [34]. In addition, GCIs are found in the white matter and subcortical regions such as the striatum, thalamus, and substantia nigra. At the clinical level, the cases affected by this type of TDP-43 pathology generally present with behavioral variant of frontotemporal dementia (bvFTD) or β-amyloid protein experimental model (APPnfv).
2.2. FTLD-TDP Type B
Type-B FTLD-TDP cases show NCI lesions in the outer and deep cortical layers with relatively little DN or NII [34]. Immunohistological analyzes also reveal a significant number of pre-inclusions and neurites, mainly in the superficial layer of the cortex. The presence of NCI lesions in lower motor neurons is also one of the specificities of type B, even in the absence of clinical signs of ALS. Glia is also affected, with many GCIs in the medullary white matter and the spinal cord—most cases have an FTLD/ALS clinical phenotype present with FTLD-TDP type B.
2.3. FTLD-TDP Type C
Type-C FTLD-TDP is characterized by long neuritic lesions, mainly in the outer layer of the cortex [34]. Some NCI lesions can also be found, both in the cortex and in the hippocampus. The presence of NII and GCI is sporadic. Type C is the most common pathologic subtype in patients with semantic variant primary progressive aphasia (svPPA).
2.4. FTLD-TDP Type D
This pathologic subtype’s hallmark is abundant lentiform NII lesions and short neuritic lesions in the neocortex [34]. Various subcortical regions (basal ganglia, thalamus, hippocampus, cerebellum, etc.) can also be heterogeneously affected. This histopathological presentation is rare since it is only found in cases carrying a mutation in the valosin-containing protein (VCP) gene.
The etiology of most cases of FTLD and ALS is still unclear; yet, these cases are related to each other by the presence of TDP-43 pathology characterized by cytoplasmic delocalization, presence of truncated forms, aggregation, and post-translational modifications of this protein (Figure 2).

Figure 2. Histopathological subtypes of FTLD-TDP. Type A is characterized mainly by short dystrophic neurites (DNs) and neuronal cytoplasmic inclusions (NCIs) in the superficial neocortical layers. Type B is characterized by NCI in the superficial and deep neocortical layers. Type C is characterized by long DN lesions in the superficial layers. Type D is characterized by lentiform neuronal intranuclear inclusions (NIIs). Glial cytoplasmic inclusion (GCI) presents in white matter only in types A and B. Created with Biorender.com.
3. FTLD-TDP Genetics
3.1. Granular Precurson (GRN) Gene
The first mutations in the GRN gene were demonstrated in 2006. More than 69 pathological mutations in the progranulin gene have been identified [35][65], representing a number of cases from 5 to 20% of FTLD of familial origin. Most of the mutations affecting the GRN gene are of the nonsense type and lead to reduced gene expression, suggesting that haploinsufficiency is the primary pathological mechanism. Carriers of these mutations show a 50% reduction in the amount of mRNA and around 33% reduction in protein.
3.2. Progranulin (GRN) Structure
Like microtubule-associated protein tau (MAPT), the GRN progranulin gene is located on chromosome 17 at position q21. This gene encodes a 68.5 kDa protein composed of 593 cysteine-rich amino acids. The full-length form of the protein contains 7.5 well-conserved domains, each consisting of a repeat of 12 cysteine motifs (also called granulin motifs) separated by junction regions. Once in the extracellular medium, various proteases can cleave progranulin at the level of these regions to generate fragments ranging from 6 to 25 kDa, called granulins [36][66].
3.3. Expression in the CNS of Progranulin
Progranulin is a secreted protein expressed in many tissues and cell types throughout the body. In the brain, its expression is low during the early stages of development and increases with age. Regarding its distribution in different cell types, progranulin is found mainly in neurons and microglia, although it may be present in small amounts in astrocytes and ependymocytes [37][67]. Its expression within microglia is significantly increased under pathological conditions [38][39][68,69].
3.4. Modulation of Expression by Transmembrane Protein 106B (TMEM106B)
The penetrance of mutations in the GRN gene is incomplete, so knowing the factors that regulate the expression of this gene is essential from a therapeutic point of view. A genome-wide association study (GWAS) of FTLD-TDP cases identified the gene encoding TMEM106B as a modulator of pathology in patients with or without a GRN gene mutation. The genetic variability of TMEM106B notably affects the penetrance of the disease in cases carrying a mutation in the GRN gene, but also the age of onset of the first symptoms [40][70]. In addition, people with the risk allele show a lower plasma level of progranulin [40][41][70,71]. The TMEM106B gene encodes a transmembrane protein capable of associating with progranulin in endolysosomes [42][72]. Regarding the underlying mechanism, a study in cells indicates that the increase in the expression of TMEM106B promotes the intracellular accumulation of progranulin [43][73]. This would imply the sequestration of progranulin within the lysosomes, which would block its release into the extracellular space and/or reduce its degradation.
In summary, the genes that are associated with FTLD-TDP are GRN, MAPT, C9ORF72, TARDBP, and VCP, although other genes not yet properly identified for FTLD-TDP but that may be related are HNRNPA2B1, SQSTM1, UBQLN2, and TREM2 [44][74], as can be seen in Table 1.
Table 1.
Summary of mutations associated with FLTD-TDP.
| Mutation | Gene | Protein | Reference |
|---|---|---|---|
| C9ORF72 expansion | C9ORF72 | TDP-43 | [45][46][47][75,76,77] |
| GRN mutation | GRN | Progranulin | [48][49][50][78,79,80] |
| MAPT mutation | MAPT | Tau | [51][52][81,82] |
| TBK1 mutation | TBK1 | TANK-binding kinase 1 | [53][83] |
| VCP mutation | VCP | Valosin-containing protein | [54][55][56][84,85,86] |
| CHMP2B mutation | CHMP2B | Charged multivesicular body protein 2B | [57][58][87,88] |
| SQSTM1 mutation | SQSTM1 | Sequestosome-1 (p62) | [59][60][89,90] |
| TIA1 mutation | TIA1 | TIA-1 RNA-binding protein | [61][91] |
3.5. Biological Function of Progranulin
Progranulin involves many physiological and pathological mechanisms, including cell proliferation, inflammation, and metabolic diseases. On the other hand, its role in the brain remains largely unknown despite intense research since the discovery of its involvement in FTLD in 2006.
3.6. Progranulin Neuronal Function and Growth of Neural Tracts
Regarding the role of progranulin as a growth factor at the peripheral level, many studies have focused on the potential role it plays in neuronal growth and survival. The expression of progranulin or granulin (E) has been shown to promote neuronal survival and neurite outgrowth in rat primary neuronal cultures [62][92]; furthermore, a study in zebrafish underscores the vital role of progranulin in the development of motor neurons. Repression of the expression of progranulin homologs in zebrafish induces a reduction in axonal growth. In murine primary neuron cultures, treatment with progranulin shows an increase in the development of neuronal tracts via glycogen synthase kinase-3beta (GSK-3β). In contrast, using small interfering RNA (siRNA) directed against progranulin decreases the neurite tree in primary cultures of rats [63][93]. The addition of recombinant progranulin in GRN -/- mice has been shown to restore neurite outgrowth. In a progranulin-deficient mouse model, analysis of dendrite size at the CA1 pyramidal cell level (hippocampus) once again supports the role of progranulin in neurite outgrowth: given the low expression of progranulin during embryonic development, its role in neuritic growth is probably more associated with synaptic plasticity in the adult brain (Figure 3).
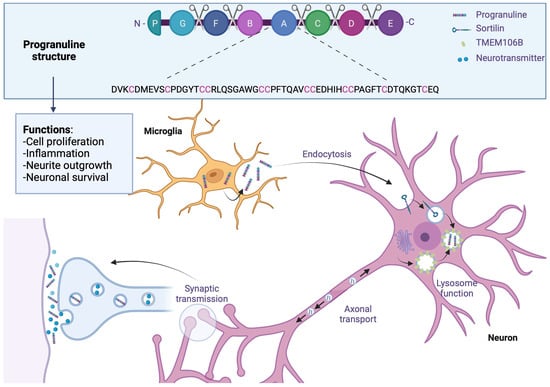
Figure 3. Progranulin in frontotemporal lobar degeneration. Progranulin contains 7.5 conserved domains, consisting of a repeat of 12 cysteine motifs separated by junction regions where some proteases will be able to cleave progranulin. Progranulin is found mainly in neurons and microglia, where its expression is increased in FTLD; through endocytosis, progranulin is bound to the sortilin receptor and sequestrated in lysosomes which are associated with TMEM106B, which would block its release into extracellular space and reduce its degradation. In adult brains, progranulin is associated with synaptic plasticity; patients with progranulin gene mutation reported an increase in synaptic vesicles and extrasynaptic secretion. Created with Biorender.com.
3.7. Synaptic Plasticity
Evidence suggests that progranulin can modulate synaptic biology. Thus, an increase in the number of synaptic vesicles and their probability of secretion has been observed in cultures of rat hippocampal neurons deficient in progranulin. These results were confirmed in the brains of FTLD patients with a mutation in the GRN gene [63][93]. Although another study indicates the opposite by highlighting a decrease in the number of these vesicles in GRN -/- mice, all these data support the role of progranulin in synaptic function. In addition, dysregulation of miRNA expression targeting proteins involved in synaptic biology was demonstrated in FTLD-PDD patients carrying a GRN gene mutation [64][94]. In addition, the stimulation of neuronal activity would promote the recruitment of progranulin at the synaptic level and its synaptic and extrasynaptic secretion. A link between loss of progranulin and synaptic dysfunction has been described; in fact, a study has shown an increase in inhibitory synapse removal in mice whose progranulin expression has been repressed in microglia. This phenomenon would be mediated in particular by increased complement expression resulting from increased microglial activation in humanized immune system (HIS) mice [65][95]. Progranulin can also bind to the sortilin receptor, which is known to bind neuropeptides such as neurotensin and neurotrophic factor pro-nerve growth factor (NGF). Progranulin binding to the sortilin receptor causes its endocytosis and rapidly leads to its degradation via the lysosomal pathway. However, no signaling pathway appears to be activated [66][67][96,97], suggesting only a role for sortilin in regulating extracellular progranulin concentration [68][98]. Progranulin would also be involved in the stress response, which would play a protective role (Figure 5).
3.8. Microglial Functions
Due to the overexpression of progranulin in pathological conditions, several studies have focused on its role in microglia. Thus, a study observed an excessive secretion of cytokines in primary cultures whose microglial progranulin expression is repressed [69][99]. Furthermore, this excess of cytokines appears to be cytotoxic. A study conducted in a similar mouse model also showed an increase in complement production [65][95]. In contrast, using siRNAs directed against progranulin decreases cytokine production in human fetal microglial cells after lipopolysaccharide (LPS) stimulation [70][100]. Progranulin also seems to act as a chemoattractant for microglial cells, suggesting a role in the recruitment of these cells during CNS damage.
3.9. Progranulin on TDP-43 Aggregation
Transactive response DNA-binding protein pathology is characterized by phosphorylated forms of the complete TDP-43 protein and C-terminal fragments in the brain tissue of FTLD patients. The relationship between progranulin haploinsufficiency and the development of this pathology is unknown. Although several studies in vitro observe proteolysis and aggregation of TDP-43 during progranulin depletion [71][72][73][56,101,102], these results are still controversial. Furthermore, no pathological modification of TDP-43 is found after the repression of the progranulin gene in zebrafish or human cell lines [74][103]. Progranulin-deficient mouse models show highly variable phenotypes; thus, while some models are distinguished by TDP-43 phosphorylation and/or delocalization [75][76][104,105], others do not show signs of TDP-43 pathology, and this occurs even at older ages. However, a recent study demonstrated that certain granulins, particularly granulin (E), would promote TDP-43 toxicity in different animal models. An accumulation of this fragment is also observed in brain regions of patients affected by the TDP-43 pathology [77][106].
3.10. Neuropathology and Associated Clinical Signs
The onset of symptoms is later than for MAPT mutations, with a mean between 59 and 65 years. However, the duration of the evolution is similar, with an average of 9 years. Singularly, there is significant heterogeneity in the clinical symptoms observed in patients, even in individuals with identical mutations or belonging to the same family. Most cases present with clinical signs of bvFTD or svPPA, often accompanied by Parkinsonian syndrome. An association with ALS is found very rarely [78][107]. The main feature observed on imaging in patients carrying a GRN mutation is the presence of asymmetric brain atrophy [79][108].
3.11. White Matter Abnormalities
Severe cortical atrophy and parietal lobe involvement are also found in these patients. From a neuropathological point of view, TDP-43 type-A inclusions are mainly found. Inclusions can also appear as NII and are primarily located in the frontal cortex and striatum [80][109]. Dystrophic neurites are frequently found in the superficial cortical layers, and minor TDP-43-positive neurites are found in the CA1 hippocampus [81][82][110,111]. Furthermore, morphometric studies indicate that the TDP-43 pathology affects specific neuronal circuits in patients with a progranulin mutation. The asymmetric distribution of TDP-43 lesions, distributed mainly in the left hemisphere, makes it possible, in particular, to differentiate TDP-43 pathology related to GRN mutations from that sometimes found in patients with AD (Figure 4).
Figure 4. Alterations due to TDP-43 pathology. TDP-43 pathology induces mutations that trigger important changes in progranulin. In addition, inclusions by TDP-43 can alter brain morphology. Created with Biorender.com.
3.12. Nucleotide C9ORF72
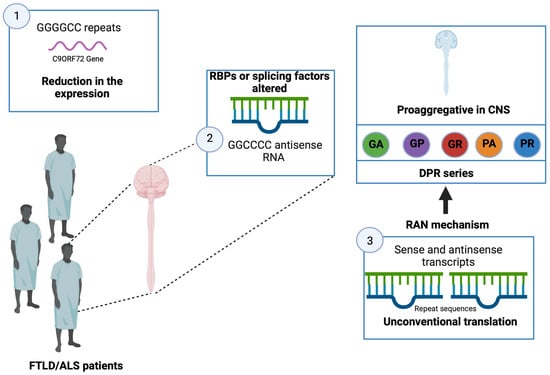
In 2011, a GGGGCC hexanucleotide repeat was identified in a non-coding region of the C9ORF72 gene as the major common genetic cause of FTLD and ALS [83][112]. In fact, although variable, the expansion size is approximately 23 repeats in a healthy individual, while it can reach several thousand repeats in affected patients [83][112]. Although the exact minimum number of repeats to induce pathology is still not well determined, most studies consider the presence of more than 30 repeats pathological [83][84][112,113]. Currently, 20 to 40% of familial cases of ALS and FTLD, respectively, are explained by a C9ORF72 mutation [85][114]. Given the significant involvement of C9ORF72 in FTLD and ALS, research has focused on understanding how the GGGGCC repeats lead to the phenomenon of neurodegeneration.
3.13. Pathophysiological Mechanisms Related to GGGGCC Repeats
Three hypotheses are proposed to explain hexanucleotide repeats’ pathological mode of action.
- The presence of many repeats could cause a reduction in the expression of the C9ORF72 gene leading to a loss of its physiological function. It would seem that the expression of the C9ORF72 gene is reduced in patients carrying this type of mutation
- [
- ]
- [
- ]
- .
- It has been shown that brain and spinal cord tissue from FTLD/ALS patients is not distinguished by the presence of nuclear foci composed of GGGGCC RNA but also GGCCCC antisense RNA
- [
- ]
- [
- [
- ]
- ,
- ]
-
The transcripts (sense and antisense) produced from the repeated sequences would be the target of an unconventional translation mechanism that does not depend on the presence of an ATG codon. This mechanism, called translation-associated repeat not initiated by ATG (RAN), would be responsible for the production of a series of dipeptides (dipeptide repeat, DPR; glycine-alanine, GA; glycine-proline, GP; glycine-arginine, GR; proline-alanine, PA; and proline-arginine, PR) [87][116]. These DPRs, located throughout the entire CNS, have the characteristic of being pro-aggregative, and, therefore, could participate in the neurodegenerative process [87][116]. Several studies carried out in cell culture models of Drosophila demonstrate the toxicity of these DPRs [86]115[89][,118] (Figure 5).
Figure 5. Alterations in patients with FTLD/ALS. In patients with FTLD/ALS, a reduction in the expression of the C9ORF72 gene is present, and alteration in RBPs as well as in splicing factors and changes in transcription processes are observed. Created with Biorender.com.