The epithelial-mesenchymal transition (EMT) is a complicated biological process in which cells with epithelial phenotype are transformed into mesenchymal cells with loss of cell polarity and cell–cell adhesion and gain of the ability to migrate. EMT and the reverse mesenchymal-epithelial transitions (METs) are present during cancer progression and metastasis. Using the dynamic switch between EMT and MET, tumour cells can migrate to neighbouring organs or metastasize in the distance and develop resistance to traditional chemotherapy and targeted drug treatments. Growing evidence shows that reversing or inhibiting EMT may be an advantageous approach for suppressing the migration of tumour cells or distant metastasis. Among different levels of modulation of EMT, alternative splicing (AS) plays an important role.
- epithelial-mesenchymal transitions
- alternative splicing
- cancer
1. Introduction
2. The Epithelial-Mesenchymal Transition
2.1. Mechanism of EMT
The main features of EMT are downregulation of epithelial cell characteristics and acquisition of mesenchymal properties (Figure 1). The first step of EMT is a disassembly of the epithelial cell–cell contacts, which include the tight junction, adherens junction, desmosomes, and gap junction, as well as the loss of cell polarity. The key cell–cell adhesion molecules—epithelial cadherin (E-cadherin) and cytokeratin—are downregulated while neural cadherin (N-cadherin), vimentin, and fibronectin, are upregulated. Cells acquire the mesenchymal phenotype with motility and invasive capacities by forming lamellipodia, filopodia, and invadopodia, followed by expressing matrix metalloproteinases which have the ability to degrade extracellular matrix proteins [7].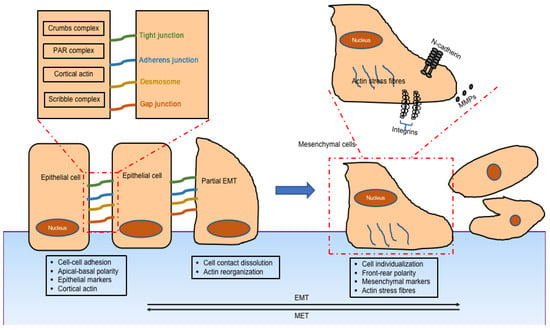
2.2. Classification of EMT
Type 1 EMT (Figure 2A) is encountered during embryogenesis. The primitive epithelium transforms to the primary mesenchyme through EMT, then the primary mesenchyme is re-induced to generate secondary epithelia through the reverse MET. These secondary epithelia can then develop into diverse kinds of epithelial tissues. It is related to embryonic development, such as implantation, embryonic gastrulation, and organ development.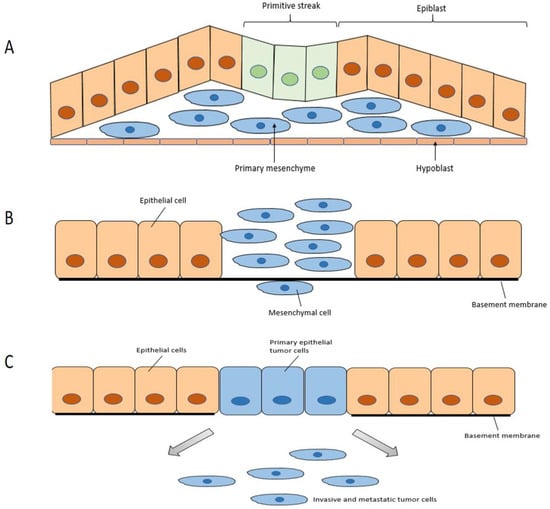
2.3. Regulation of EMT
The mechanisms regulating EMT are quite complex. Mechanical signals and biochemical signals are both playing important roles in this regulation. There are many regulators at several different molecular levels, including transcriptional and post-transcriptional levels like mRNA processing or microRNAs [1]. Snail family zinc-finger transcription factor (SNAIL) is one of the most important transcription factors (TFs) involved in EMT; it was first found as a transcriptional repressor of E-cadherin and originally identified as a repressor of E-cadherin homologs in Drosophila [10]. Proteins within the SNAIL family in vertebrates have been subdivided into two subfamilies: Snail1 (known as SNAI1) and Slug (known as SNAI2) [11]. They both can induce EMT by directly inhibiting the transcription of E-cadherin [12]. Twist family BHLH transcription factor (TWIST) is a basic helix-loop-helix protein which is an important TF for EMT during metastasis and embryonic morphogenesis [13][14]. TWIST represses E-cadherin and induces EMT and cell migration by directly or indirectly suppressing the E-cadherin transcription via target E-boxes in the E-cadherin promoter [14][15]. Zinc-finger E-box binding homeobox (ZEB) family of TFs, similar to SNAIL and TWIST, bind the E-boxes and repress epithelial junctions and polarity genes as well as activate mesenchymal genes during EMT [15][16]. Two subfamilies of proteins from the ZEB family in vertebrates have been reported: ZEB1 and ZEB2. They can both inhibit or stimulate the transcription process by attaching to E-boxes on regulatory gene sequences [16][17]. MicroRNAs (miRNAs) are also key regulators of epithelial phenotype during EMT. They can regulate E-cadherin expression either directly or indirectly and regulate the expression of TFs during EMT, as well as other target genes, to assist in identifying the epithelial or mesenchymal phenotype [17][18]. Additionally, long-noncoding RNA (lncRNAs) as a post-transcriptional regulator are also involved in EMT regulation, while miRNAs regulate EMT-TF [17][18][19][18,19,20].3. Alternative Splicing
3.1. Splicing
The basic process of splicing contains two major steps (Figure 3): spliceosome assembly and pre-mRNAs actual splicing. These two processes are catalysed by the cooperation of five core small nuclear ribonucleoprotein (snRNP) particles (U1, U2, U4, U5 and U6) and numerous auxiliary proteins. The spliceosome is a macromolecular ribonucleoprotein complex composed of many proteins and small nuclear RNA molecules [20][26]. The process of spliceosome assembly is directed by consensus sequences at splice sites and results in the sequential binding and release of snRNPs, protein factors, and the disruption and formation of RNA–RNA, RNA–protein, and protein–protein interactions. Pre-mRNA splicing is a process of removing introns from pre-mRNA through two consecutive transesterification reactions and the ligation of exons in the mRNA [21][27].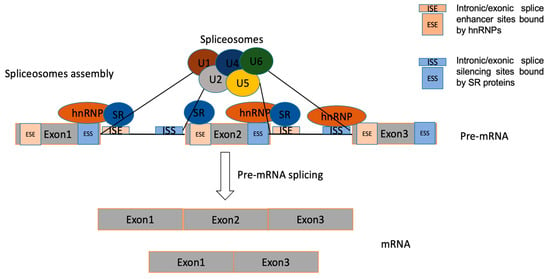
3.2. Basic Modes of Alternative Splicing
There are five major types (Figure 4) [22][28]: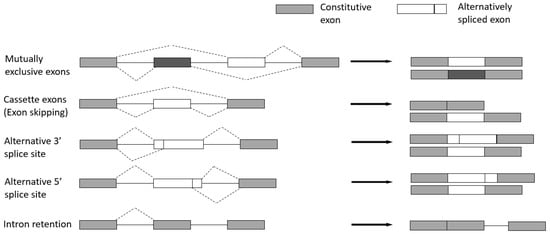
-
Cassette exons (Exon skipping)—the most prevalent pattern in which exons are spliced out of the gene or retained in the transcript.
-
Mutually exclusive exons—only one of two consecutive exons is retained in the mature transcript.
-
Alternative 3′ splice site—when the splice junction at the 3′ end is changed.
-
Alternative 5′ splice site—when the splice junction at the 5′ end is changed. Both alternative 3′ and 5′ splice sites can cause changes in the coding sequence.
-
Intron retention—when an intron is retained in the final transcript.
3.3. Regulation of Alternative Splicing
The regulation of AS is a highly dynamic combinatorial process that relies on complex coordination between intracellular and post-transcriptional processes [23][29]. Regulation of AS can be triggered by the interaction of multiple RNA-binding proteins (RBPs), which bind to cis-regulatory elements around the splice sites resulting in the utilization of the regulated splice site being enhanced or repressed [20][26]. The cis-elements, which can be located in either exons or introns, are bound by regulatory proteins to enhance or silence splicing of adjacent regulated exons, to therefore determine whether the mature transcript includes or skips certain exons [24][30]. Splicing regulatory elements can be divided into four categories: exonic/intronic splicing enhancer/silencers [25][31]. Precise control of AS is vital for normal cells, as aberrant expression of splicing is a common cause of diseases including cancer. The trans-acting splicing factors belong mostly to two large families—SR proteins, which normally bind to splicing enhancers and promote spliceosomes assembly, and hnRNPs opposing SR protein function, which generally bind to splicing silencers to inhibit exon recognition and promote exon skipping [26][27][32,33].
4. Manipulation of Alternative Splicing in EMT as a Potential Therapy for Cancer
4.1. Switching Specific Alternative Splicing Patterns in EMT
There are a series of alternative spliced events in EMT which play essential roles during cancer progression. Previous studies show that changing the splicing patterns of these gene isoforms can change phenotypes in both EMT and cancer. Thus, targeting splicing patterns is a potential method of cancer management.4.1.1. Fibroblast Growth Factor Receptor 2 (FGFR2)
FGFR2 is one of the well-recognized genes spliced differently in EMT which is essential in embryogenesis and organ regeneration [28][29][30][31][46,47,48,49]. The FGFR family functions through the binding of FGF ligands to their receptors, activation via dimerization and, therefore, prompting the tyrosine kinase domains to a set of subsequent signal activations [32][50]. Following this cell division, growth and differentiation are induced [33][34][35][36][51,52,53,54]. There are two mutually exclusive FGFR2 splice isoforms—FGFR2 IIIb and FGFR2 IIIc. Splicing switch from FGFR2 IIIb to FGFR2 IIIc has been implicated in pathological EMT [37][55]. Epithelial phenotype involves exon IIIb inclusion while, in EMT, exon IIIc inclusion induces a mesenchymal phenotype. It has been shown that ESRP1 and 2 are regulating splicing of FGFR2, switching to the epithelial phenotype—FGFR2 IIIb isoform [38][56]. ESRP expression is higher in epithelial cells and decreases in EMT. The switch from exon IIIc to IIIb of FGFR2 in mesenchymal cells may be promoted by ESRP overexpression.4.1.2. Recepteur d’Origine Nantais (RON, MST1R)
The proto-oncogene RON, also known as macrophage stimulating 1 receptor (MST1R), is a member of the receptor tyrosine kinase (RTK) family. The mature RON protein is a 180 kDa heterodimer composed of a 40 kDa α chain and a 150 kDa β chain. The transmembrane β chain has tyrosine kinase activity, and the precursor (pro-RON) exists as a single chain [6]. The extracellular sequence of RON includes amino-terminal semaphoring (Sema), plexin-semaphorin-integrin (PSI), and four immunoglobulin-like IPT domains. Variants are mainly generated through mechanisms such as alternating pre-mRNA splicing, protein truncation and alternative transcription. RON and its alternatively spliced variants, including RONΔ85, Δ110, Δ155, Δ160, Δ165, and Δ170, play essential roles in numerous tumour biological activities in cancer, such as cell–cell adhesion, proliferation, apoptosis, and EMT [39][59]. The production of the ΔRon165 isoform during EMT is generated by exclusion of exon 11, which results in lacks of 49 amino acids (aas) in the extracellular β-chain [6].4.1.3. CD44
The protein encoded by CD44 is part of a group of integral membrane glycoproteins which mediate cell–cell interactions and the interaction between cells and the extracellular matrix, together with cell adhesion and migration. The CD44 gene transcript undergoes complex alternative splicing, resulting in many functionally different protein subtypes. These different isoforms have diverse tissue-specific functions and are involved in various cellular processes, including tumour progression and metastasis. It has been found that expression of CD44v (CD44 novel splice variant) isoforms is often associated with initiation, progression, and metastasis of colon, prostate, intestinal, gastric, and breast cancers [40][41][42][43][44][45][72,73,74,75,76,77].4.1.4. Catenin Delta 1 (CTNND1)
The CTNND1 gene encodes a member of the Armadillo protein family, also known as p120, which plays a role in both oncogenic and tumour suppressor functions, and its alternative splicing is related to cell proliferation, migration, invasion, and EMT [46][47][48][81,82,83]. Two isoforms of CTNND1 generated by AS, p120-1 and p120-3, are associated with distinctive functions and different interactions in various cell types. A long mesenchymal-specific splice variant with exons 2 and 3, p120-1 is normally mostly expressed in mesenchymal cells, whereas p120-3 is a shorter isoform with lack of these exons and is mostly predominant in epithelial cells [49][50][84,85]. p120 isoform expression evaluation within a panel of breast cancer cell lines revealed that more invasive cells show higher expression of the large isoforms p120-1 and p120-2; however, p120-3 is expressed in all cell lines [51][86]. As p120-3 often has high expression levels in epithelial cells, a switch from isoform p120-1 to p120-3 might be a possible way to revise EMT and inhibit tumour growth.4.1.5. Other Genes Spliced Differently during EMT
ARHGEF11 (Rho guanine nucleotide exchange factor 11) is the guanine nucleotide exchange factor (GEF) for the RhoA small GTPase protein. It has been revealed to promote tumour metastasis in glioblastoma and ovarian carcinoma and promotes proliferation and EMT of hepatocellular carcinoma by activating β-catenin [52][87].
CCND1 has two isoforms derived by splicing, named cyclin D1a and cyclin D1b, with inclusion of intron 4 in D1b mRNA. Both D1a and D1b are often upregulated in human cancers; however, cyclin D1b alone can induce cellular transformation, and is correlated with cancer progression and poor prognosis in prostate, colon, colorectal, and urinary bladder cancer [53][54][55][56][57][89,90,91,92,93]. Androgen receptor (AR) variants, especially AR3, could contribute to prostate cancer progression through inducing EMT, achieving stem cell characteristics, and regulating stem cell related pathways [58][59][60][94,95,96]. Research revealed that the expression of MBNL1 isoforms lacking exon 7 (MBNL1 Δex7) proteins plays a role as a tumour suppressor, as cancer cells tend to downregulate in the presence of MBNL1 isoforms containing exon 7 [61][97]. Zinc-finger antiviral protein (ZAP) is an important antiviral factor that specifically inhibits the replication of a variety of viruses by binding to the target RNA sequence of the virus and interfering with the translation initiation of the target mRNA. ZAP has two major isoforms that result from alternative splicing at their C terminus: ZAPL (long) encodes a poly (ADP-ribose) polymerase (PARP)-like domain that is missing from ZAPS (short).