Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Kiyofumi Yamada and Version 1 by Kiyofumi Yamada.
Schizophrenia is one of the most serious psychiatric disorders and is characterized by reductions in both brain volume and spine density in the frontal cortex. RhoA belongs to the RAS homolog (Rho) family and plays critical roles in neuronal development and structural plasticity via Rho-kinase. RhoA activity is regulated by GTPase-activating proteins (GAPs) and guanine nucleotide exchange factors (GEFs). Several variants in GAPs and GEFs associated with RhoA have
been reported to be significantly associated with schizophrenia. Here, we summarize clinical evidence showing that variants in genes regulating
RhoA activity are associated with schizophrenia.
- copy number variants
- CNVs
- single-nucleotide polymorphisms
1. Introduction
Schizophrenia is one of the most serious psychiatric disorders and affects approximately 1% of the population [1]. It typically emerges in late adolescence and early adulthood and involves positive symptoms (such as hallucinations, delusions, and formal thought disorder), negative symptoms (such as lack of volition, reduced speech output, and flattening of affect), and cognitive dysfunction (manifested, for instance, by deteriorations in working memory, executive function, and learning) [1][2][3].
Neuropathological and neurophysiological changes observed in patients with schizophrenia include enlargement of the lateral ventricles and a 2% decrease in gray matter volume [4]. Brain volume reduction involves the frontal lobe in particular, including the frontal cortex, which exhibits a reduced density of pyramidal neuron spines that are components of the postsynaptic site of most excitatory synapses [4][5][6][7][8][9]. Moreover, patients with schizophrenia show decreased prefrontal cortex (PFC) blood flow during the performance of cognitive tasks [10].
One of the major therapeutic targets of schizophrenia is the dopamine D2 receptor, and its antagonists, such as haloperidol, reduce positive symptoms but have a minimal effect on negative symptoms or cognitive deficits [1][11]. These drugs also have major side effects, including sedation, hyperprolactinemia, and the extrapyramidal symptoms of parkinsonism [1]. Second-generation antipsychotics such as risperidone and olanzapine have lower rates of such severe side effects, but their clinical efficacy and tolerability are not significantly improved [12]. Furthermore, 20–30% of patients show resistance to antipsychotic treatment [13]. Clozapine is the sole drug indicated for treatment-resistant schizophrenia and improves symptoms in only about 30–60% of patients [14][15][16][17]. Therefore, there is an urgent need for the development of effective schizophrenia treatments that are both effective and safe. To achieve this goal, it is necessary to understand the pathoetiology of the disease and to establish novel pathophysiologic animal models to expand upon existing classical pharmacologic animal models.
The etiology of schizophrenia involves both genetic vulnerabilities and environmental risk factors such as pregnancy and birth complications, childhood trauma, substance abuse, and psychosocial stress in adolescence [1][18]. In genome-wide association studies (GWASs) of schizophrenia, more than 200 genetic loci associated with neuronal function, including synaptic organization, differentiation, and transmission, have been shown to be associated with schizophrenia [19]. In addition to common variants, a small number of rare copy number variants (CNVs) and gene-disrupting variants, including the so-called rare-coding variants and protein-truncating variants, have been identified in schizophrenia with large effect sizes (odds ratios (ORs) of 2–60 fold and 3–50 fold, respectively) [1]. Several CNVs, such as 1q21.1, 2p16.3 (NRXN1), 3q29, 15q11.2, 15q13.3, and 22q11.2 have been consistently reported to be associated with schizophrenia [20][21]. Furthermore, a gene set analysis replicated previous findings (e.g., those implicating synapses and calcium signaling) and identified novel biological pathways such as those involved in the oxidative stress response, genomic integrity, and kinase and small GTPase signaling [22].
The Rho GTPase family plays a role in spine morphology by regulating actin dynamics [23]. It is associated with psychiatric diseases such as schizophrenia and depression, and also with neurodevelopmental disorders including autism spectrum disorders and intellectual disabilities [24][25][26][27][28][29][30][31]. Variants in genes upstream of the Rho GTPase family, such as KALRN and p250GAP, have been reported to be associated with schizophrenia [25][27].
2. Rho Family Activity Is Regulated by GTPase-Activating Proteins (GAPs) and Guanine Nucleotide Exchange Factors (GEFs)
RhoA belongs to the RAS homolog (Rho) family, along with cell division control protein 42 (Cdc42) and RAS-related C3 botulinum toxin substrate 1 (Rac1) [32]. Rho family proteins contain a conserved GDP/GTP binding domain and switch their activity by cycling between GDP-bound (inactive) and GTP-bound (active) states [32][33]. This cycling is regulated by GAPs, GEFs, and guanine nucleotide dissociation inhibitors (GDIs) [32][34]. GAPs consist of more than 70 members, and conversion from a GTP-bound form to a GDP-bound form suppresses their activity [32][35]. In contrast, GEFs (>74 members) accelerate the exchange of tightly bound GDP for GTP, resulting in the activation of Rho family proteins [32][35]. GAPs and GEFs exhibit high selectivity for RhoA, Cdc42, and Rac1 [35][36]. GDIs, of which there are only three members in the human genome, form soluble complexes with GDP-bound Rho protein and control its cycling between the cytosol and membrane [32][34].3. Rho Family Protein Effectors and Their Physiological Roles in the Brain
Rho family proteins are associated with over 70 potential effector proteins [37]. Rho-kinase, a serine/threonine kinase, is a representative downstream effector of RhoA [38]. In vascular smooth muscle, for example, Rho-kinase phosphorylates myosin phosphatase-targeting subunit 1 (MYPT1) at Thr696 and Thr853. This converts MYPT1 to an inactivated state, increases the phosphorylation of myosin light chain, and promotes actomyosin contractility [39][40][41]. P21-activated kinase (PAK) acts as a downstream effector for Cdc42 and Rac1 and affects actin dynamics by regulating the LIM kinase–cofilin pathway [42][43]. PAK also inhibits myosin light chain kinase, resulting in decreased myosin light chain phosphorylation and, thus, decreased actomyosin contractility [44]. Rho GTPases regulate cell morphology. For instance, RhoA promotes stress fiber formation and focal adhesions in cells [45]. Rho GTPases also modulate neuronal development. For instance, RhoA inhibits growth of dendrites and axons, while Rac1 and Cdc42 promote axonal elongation [42][46]. In addition, RhoA/Rho-kinase signaling promotes spine shrinkage and destabilization, while Rac1 and Cdc42/PAK signaling promotes spine stabilization and maintenance [23][47]. Accordingly, Rho GTPase signaling is involved in neuronal maturation through the regulation of actin dynamics.4. Schizophrenia-Associated Genes Involved in Small GTPase RhoA Signaling
Recently, several variants of RhoA-associated GAPs, including ARHGAP10 [48], ARHGAP18 [49][50][51], and p250GAP [52], and GEFs such as KALRN [53][54][55][56] and ARHGEF11 [57], were reported to be significantly associated with the development of schizophrenia. In addition, variants in genes that activate RhoA via GEF, such as RTN4R [58][59][60], or in those that degrade RhoA, such as KCTD13 [61], were also identified in schizophrenia (Figure 1).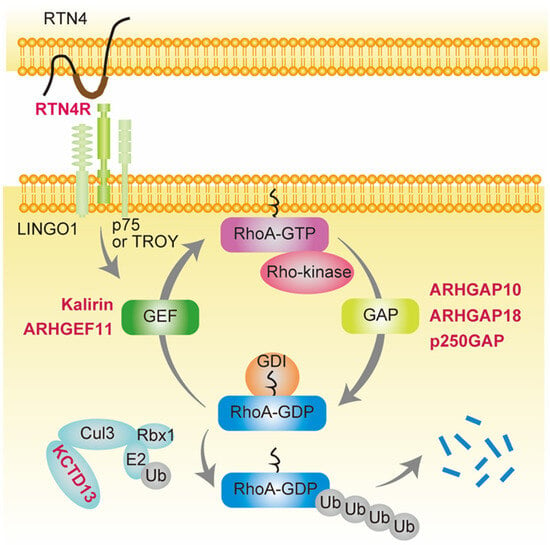
Figure 1. Schizophrenia-associated genes involved in small GTPase RhoA signaling. Genes shown in red are schizophrenia-associated genes involved in small GTPase RhoA signaling. RhoA contains a conserved GDP/GTP binding domain, and its activity cycles between GDP-bound (inactive) and GTP-bound (active) states. ARHGAP10, ARHGAP18, and p250GAP are GTPase-activating proteins (GAPs) that convert RhoA from the GTP- to GDP-bound form, thereby suppressing its activity. In contrast, Kalirin and ARHGEF11 are guanine nucleotide exchange factors (GEFs) that accelerate the exchange of tightly bound GDP for GTP, resulting in RhoA activation. GDIs form soluble complexes with GDP-bound RhoA and control its cycling between the cytosol and membrane. Reticulon 4 receptor (RTN4R) (also called Nogo-66 receptor, NgR1), a RTN4 receptor subunit, is activated by RTN4 and binds leucine-rich repeat and immunoglobulin domain-containing protein (Lingo-1) and either the p75 neurotrophin receptor or tumor necrosis factor (TNF) receptor orphan Y (TROY), resulting in RhoA activation by GEF. Potassium channel tetramerization domain-containing 13 (KCTD13) is the only identified signaling protein capable of inducing the microcephaly phenotype associated with 16p11.2 duplication, which is associated with schizophrenia [61][62]. KCTD13 is functionally related to cullin 3 (Cul3), a core component of E3 ubiquitin-protein ligase complexes that mediates the ubiquitination and subsequent proteasomal degradation of target proteins such as RhoA.
4.1. GAPs
4.1.1. ARHGAP10
The ARHGAP10 gene, which encodes Rho GTPase-activating protein 10 (ARHGAP10), is located on chromosome 4q31.23 and exhibits GAP activity for RhoA and Cdc42 [32][63]. ARHGAP10 is expressed in the brain [63][64], and its mRNA levels rise in the cerebellum, striatum, and frontal cortex from E4 to P56 in mice [64]. CNVs in ARHGAP10 were identified in seven patients with schizophrenia (six with deletions and one with duplication) but not in controls, and there was a significant association of ARHGAP10 CNVs with schizophrenia in Japanese patients (OR = 12.3, p = 0.015) [48]. Most ARHGAP10 CNVs were exonic deletions at the Bin1/amphiphysin/Rvs167 domain, the RhoGAP domain, or both. The relative expression levels of ARHGAP10 mRNA in lymphoblastoid cell lines established from the peripheral blood of patients with exonic ARHGAP10 CNVs were significantly decreased compared to those in patients with schizophrenia without ARHGAP10 CNVs and in a control group [48]. One of the patients (case #5) with ARHGAP10 CNVs had a missense variant (p.S490P) in exon 17 that overlapped with the exonic deletion on the other allele [48]. ARHGAP10 p.S490P showed weaker binding to active RhoA compared to wild-type ARHGAP10, suggesting that this single-nucleotide variation (SNV) exhibits the loss of function of ARHGAP10. Of note, clinical data of these seven patients with ARHGAP10 variants showed that treatment response was poor in most individuals, including case #5 [48].4.1.2. ARHGAP18
ARHGAP18 is ubiquitously expressed throughout the body, including the brain, and shows GAP activity for RhoA but not for Rac1 or Cdc42 [65][66][67]. Through RhoA/Rho-kinase signaling, ARHGAP18 regulates cell spreading and migration and also the formation of stress fibers and focal adhesions [65]. ARHGAP18 knockdown, which leads to RhoA activation, causes significantly increased formation of stress fibers and focal adhesions in HeLa cells, while these changes are abolished by a Rho-kinase inhibitor or by dominant-negative RhoA transfection [65]. In contrast, the overexpression of wild-type ARHGAP18, but not GAP-defective ARHGAP18, suppresses the formation of stress fibers and focal adhesions in HeLa cells [65]. ARHGAP18 also contributes to cell migration [65]. The knockdown or knockout of ARHGAP18 impairs migration and cellular polarity in breast cancer cells (MDA-MB-231 or SUN-159 cells) [65][67]. Single-nucleotide polymorphisms (SNPs) in ARHGAP18 are associated with schizophrenia [49][50]. The genotypes and allelic frequencies of two SNPs, rs7758025 and rs9483050, were significantly different between patients and controls in a Chinese-Han population (genotype: rs7758025, p = 0.0002, and rs9483050, p = 7.54 × 10−6; allelic frequencies: rs7758025, p = 4.36 × 10−5, and rs9483050, p = 5.98 × 10−7). In addition, the AG haplotype in rs7758025-rs9385502 and the CG haplotype in rs11753915-9483050 were associated with schizophrenia (AG haplotype in rs7758025-rs9385502: p = 0.0012, 95% confidence interval [CI] = 0.48–0.93; CG haplotype in rs11753915-9483050: p = 9.6 × 10−6, 95% CI = 0.44–0.78) [49]. Another group reported an association between SNPs in ARHGAP18 and schizophrenia in Caucasian people [50][51]. They also performed a combined analysis with GWAS and functional magnetic resonance imaging scanning and demonstrated that these SNPs were significantly correlated with neuronal activity in the dorsolateral prefrontal cortex during a working memory task [50].4.1.3. p250GAP (ARHGAP32)
P250GAP (ARHGAP32) is expressed in the brain [68] and exerts GAP activity for RhoA but not Cdc42 and Rac1 in mouse primary hippocampus neurons [69]. P250GAP regulates spine morphology in primary hippocampus neurons and neurogenesis in Neuro-2A cells through the regulation of RhoA activity [69][70]. In addition, p250GAP interacts with the NR2B subunit of N-methyl-D-aspartate (NMDA) receptors and is involved in NMDA receptor-mediated RhoA activation [69][70]. An SNP in p250GAP (rs2298599) was shown to be associated with schizophrenia in a Japanese cohort (p = 0.00015) [52]. The minor genotype frequency was higher in patients with schizophrenia (18%) than in healthy controls (9%) (p = 0.000083) [52]. rs2298599 is located 2.9 kb downstream of p250GAP and showed no significant association with p250GAP expression levels in immortalized lymphoblasts in in silico analysis (p = 0.28) [52]. Thus, further study is needed to clarify the mechanism of the association between p250GAP and schizophrenia.4.2. GEFs
4.2.1. KALRN
Kalirin has two GEF domains, with activity targeting Rac1 (GEF1) and RhoA (GEF2), respectively [27][71]. The alternative splicing of KALRN gives rise to several isoforms, including Kalirin-4, Kalirin-5, Kalirin-7, Kalirin-8, Kalirin-9 (Kal9), and Kalirin-12 (Kal12) [27][71]. Kal9 and Kal12 contain both the GEF1 and GEF2 domains, while other isoforms contain only the GEF1 domain [27][71]. Kalirin is involved in neurite and dendritic outgrowth and in dendritic arborization in the brain [71]. Expression levels of Kal9 and Kal12 in the brain were found to be higher during early postnatal development than in adulthood [72]. Knockout of Kal9 and Kal12 by shRNA decreased the complexity of rat hippocampal neurons on days in vitro (DIV) 4 and 7 (immature neurons) [73]. In rat cortical neurons, Kal9 overexpression on DIV 2 (immature neurons) resulted in neurite elongation [72], while that on DIV 28 (mature neurons) reduced dendritic length and complexity [74]. These reports indicate that the role of Kal9 in neurons might change depending on the developmental stage. A transcriptome-wide association study in patients with schizophrenia revealed an increase in exon skipping immediately prior to the GEF2 domain in KALRN transcripts [53]. In addition, some missense variants in KALRN, such as P2255T and T1207M, showed a higher frequency in schizophrenia cases compared to control cases [54][56]. In particular, P2255T in KALRN was significantly associated with schizophrenia (OR = 2.09, p = 0.048) in a Japanese population [54]. Of note, a P2255 residue exists near the RhoA-GEF2 domain [55]. The P2255T variant in Kal9 (Kal9-P2255T) leads to highly stable Kal9 mRNA, resulting in increased protein levels of Kal9 [55], which, for instance, were detected in the auditory cortex of patients with schizophrenia by post-mortem analysis [74]. Furthermore, overexpression of Kal9-P2255T in rat primary neurons and HEK 293 cells increased RhoA activity but had no effect on Rac1 activity [55][75]. These data indicate that Kal9-P2255T increases the expression levels of Kal9, leading to the activation of RhoA. From the viewpoint of neuronal morphology, Kal9-P2255T overexpression in cortical primary neurons led to a significant reduction in proximal dendritic complexity and dendritic spine size compared to wild-type Kal9 (Kal9-WT) [55]. On the other hand, it is known that reticulon 4 receptor (RTN4R) activates Kal9 and subsequently leads to the activation of RhoA [76][77]. The RTN4R/Kal9/RhoA pathway is known to modulate neurite outgrowth [77]. Myelin-associated inhibitors such as oligodendrocyte-myelin glycoprotein (OMGp) have been identified as additional RTN4R (NGR1) ligands and these also suppress neurite outgrowth [75][78]. Pharmacologic inhibition of RhoA with the RhoA inhibitor CT04 prevented the OMGp-induced decrease in neuronal complexity [75]. The overexpression of Kal9-P2255T in cortical primary neurons made them more sensitive to OMGp and decreased both the length and complexity of dendritic arbors [75]. These results suggest that Kal9-P2255T-induced RhoA activation causes morphological changes in neurons.4.2.2. ARHGEF11
ARHGEF11, also referred to as KIAA0380 or GTRAP48, shows GEF activity for RhoA but not Rac1 or Cdc42 [79]. ARHGEF11 is expressed in the brain [80][81] and in cortical neurons, including dendrites and spines [82]. ARHGEF11 regulates glutamate transport activity by direct binding of excitatory amino acid transporter 4 [81]. The overexpression of ARHGEF11 was shown to decrease spine density in rat cortical neurons [82][83]. ARHGEF11 haplotypes such as C-C of rs6427340-rs6427339 and A-C-C of rs822585-rs6427340-rs6427339 were shown to be associated with schizophrenia (p = 0.0010 and 0.0018, respectively), but the ARHGEF11 SNPs were not [57]. The functions of ARHGEF11 haplotypes associated with schizophrenia have not been clarified. On the other hand, in situ hybridization analysis indicated that ARHGEF11 mRNA levels in the thalamus of patients with schizophrenia were higher than those in healthy controls [84]. These findings raise the possibility that ARHGEF11 activation is associated with schizophrenia pathology.4.3. Others
4.3.1. RTN4R
RTN4R (also called Nogo-66 receptor, NgR1) is a RTN4 receptor subunit located at chr22q11.2, and it has been shown that deletion of chr22q11.2 is associated with a high risk of developing schizophrenia [85]. RTN4R binds leucine-rich repeat and immunoglobulin domain-containing protein (Lingo-1) and either the p75 neurotrophin receptor or tumor necrosis factor (TNF) receptor orphan Y (TROY) and activates RhoA through Kal9; this results in the collapse of growth cones, which prevents further axonal growth and inhibits myelination [76][86]. Several SNPs in RTN4R are associated with schizophrenia [58][59]. In samples from individuals of Afrikaner origin, significant associations with schizophrenia were seen for SNP rs696880 in women (OR = 0.73, p = 0.046) and for rs701427 (OR = 1.21, p = 0.019), rs696880 (OR = 1.18, p = 0.029), and rs854971 (OR = 1.20, p = 0.021) in men [59]. Diffusion tensor imaging revealed that the SNP rs701428 was associated with white matter abnormalities in 22q11.2 deletion syndrome [87]. Other groups identified several rare missense variants in patients with schizophrenia, specifically p.R68H (rs145773589), p.R119W (rs74315508), p.R196H (rs74315509), p.D259N (rs3747073), p.R292H (rs1432033565), and p.V363M (rs149231717), and p.R292H was significantly associated with schizophrenia (OR = 3.9, p = 0.048) [58][60]. RTN4R-R292H is located in the ligand binding site, and its overexpression in E5.5 chick retinal neurons significantly decreased growth cone collapse induced by treatment with RTN4, a ligand of RTN4R, compared to that resulting from treatment with RTN4R-WT [58]. In addition, a glutathione S-transferase binding assay showed that RTN4R-R292H exhibited reduced interaction with LINGO1 compared to RTN4R-WT [58]. Although these data suggest that RTN4R-R292H has impaired function, the effect of RTN4R-R292H on RhoA signaling remains obscure. In a post-mortem brain analysis, the expression levels of RTN4R were decreased in the dorsolateral prefrontal cortex but increased in the hippocampal CA3 region of patients with schizophrenia compared to healthy controls [88]. In addition to the above, genetic variations in components of the RTN4R signaling pathway, such as RTN4 (p = 0.047 and 0.037 for rs11894868 and rs2968804, respectively) and myelin-associated glycoprotein (p = 0.034 and 0.029 for rs7249617 and rs16970218, respectively), were shown to be associated with schizophrenia [89].4.3.2. 16p11.2 CNVs and the KCTD13-Cul3-RhoA Pathway
16p11.2 microduplication was associated with schizophrenia in two large cohorts (OR = 25.8, p = 1.2 × 10−5; and OR = 8.3, p = 0.022) [61]. In a zebrafish model, potassium channel tetramerization domain-containing 13 (KCTD13) was identified as the sole signaling protein capable of inducing the microcephaly phenotype associated with 16p11.2 duplication [62]. A spatiotemporal protein–protein interaction network analysis showed that KCTD13 is functionally related to cullin 3 (Cul3) [90]. Cul3 is a core component of E3 ubiquitin–protein ligase complexes and mediates the ubiquitination and subsequent proteasomal degradation of target proteins such as RhoA, but not Rac1 and Cdc42 [91][92]. Although it is estimated that 16p11.2 microduplication is associated with decreased RhoA protein levels, organoids derived from patients with autism spectrum disorder who had 16p11.2 duplications showed RhoA activation and slightly increased KCTD13 expression [93]. Therefore, further research should analyze 16p11.2 microduplication in patients with schizophrenia.References
- Jauhar, S.; Johnstone, M.; McKenna, P.J. Schizophrenia. Lancet 2022, 399, 473–486.
- Gogtay, N.; Vyas, N.S.; Testa, R.; Wood, S.J.; Pantelis, C. Age of onset of schizophrenia: Perspectives from structural neuroimaging studies. Schizophr. Bull. 2011, 37, 504–513.
- Stępnicki, P.; Kondej, M.; Kaczor, A.A. Current Concepts and Treatments of Schizophrenia. Molecules 2018, 23, 2087.
- Haijma, S.V.; Van Haren, N.; Cahn, W.; Koolschijn, P.C.M.P.; Pol, H.E.H.; Kahn, R.S. Brain volumes in schizophrenia: A meta-analysis in over 18,000 subjects. Schizophr. Bull. 2012, 39, 1129–1138.
- Howes, O.D.; Cummings, C.; Chapman, G.E.; Shatalina, E. Neuroimaging in schizophrenia: An overview of findings and their implications for synaptic changes. Neuropsychopharmacology 2022, 48, 151–167.
- Glausier, J.; Lewis, D. Dendritic spine pathology in schizophrenia. Neuroscience 2013, 251, 90–107.
- Broadbelt, K.; Byne, W.; Jones, L.B. Evidence for a decrease in basilar dendrites of pyramidal cells in schizophrenic medial prefrontal cortex. Schizophr. Res. 2002, 58, 75–81.
- Konopaske, G.T.; Lange, N.; Coyle, J.T.; Benes, F.M. Prefrontal cortical dendritic spine pathology in schizophrenia and bipolar disorder. JAMA Psychiatry 2014, 71, 1323–1331.
- Runge, K.; Cardoso, C.; de Chevigny, A. Dendritic Spine Plasticity: Function and Mechanisms. Front. Synaptic Neurosci. 2020, 12, 36.
- Weinberger, D.R.; Berman, K.F.; Zec, R.F. Physiologic dysfunction of dorsolateral prefrontal cortex in schizophrenia: I. Regional cerebral blood flow evidence. Arch. Gen. Psychiatry 1986, 43, 114–124.
- Haddad, P.M.; Correll, C.U. The acute efficacy of antipsychotics in schizophrenia: A review of recent meta-analyses. Ther. Adv. Psychopharmacol. 2018, 8, 303–318.
- Leucht, S.; Cipriani, A.; Spineli, L.; Marvidis, D.; Örey, D.; Richter, F.; Samara, M.; Barbui, C.; Engel, R.R.; Geddes, J.R.; et al. Comparative efficacy and tolerability of 15 antipsychotic drugs in schizophrenia: A multiple-treatments meta-analysis. Lancet 2013, 382, 951–962.
- Elkis, H. Treatment-resistant schizophrenia. Psychiatr. Clin. N. Am. 2007, 30, 511–533.
- Kane, J.; Honigfeld, G.; Singer, J.; Meltzer, H. Clozapine for the treatment-resistant schizophrenic. Arch. Gen. Psychiatry 1988, 45, 789–796.
- Rosenheck, R.; Cramer, J.; Xu, W.; Thomas, J.; Henderson, W.; Frisman, L.; Fye, C.; Charney, D. A Comparison of clozapine and haloperidol in hospitalized patients with Refractory Schizophrenia. N. Engl. J. Med. 1997, 337, 809–815.
- Siskind, D.; Siskind, V.; Kisely, S. Clozapine Response Rates among People with Treatment-Resistant Schizophrenia: Data from a Systematic Review and Meta-Analysis. Can. J. Psychiatry 2017, 62, 772–777.
- Blackman, G.M.; Lisshammar, J.E.M.; Zafar, R.M.; Pollak, T.A.; Pritchard, M.M.; Cullen, A.E.; Rogers, J.M.B.; Carter, B.; Griffiths, K.M.; Nour, M.B.B.; et al. Clozapine Response in Schizophrenia and Hematological Changes. J. Clin. Psychopharmacol. 2020, 41, 19–24.
- Stilo, S.A.; Murray, R.M. Non-Genetic Factors in Schizophrenia. Curr. Psychiatry Rep. 2019, 21, 100.
- Trubetskoy, V.; Pardiñas, A.F.; Qi, T.; Panagiotaropoulou, G.; Awasthi, S.; Bigdeli, T.B.; Bryois, J.; Chen, C.-Y.; Dennison, C.A.; Hall, L.S.; et al. Mapping genomic loci implicates genes and synaptic biology in schizophrenia. Nature 2022, 604, 502–508.
- Kushima, I.; Nakatochi, M.; Aleksic, B.; Okada, T.; Kimura, H.; Kato, H.; Morikawa, M.; Inada, T.; Ishizuka, K.; Torii, Y.; et al. Cross-Disorder Analysis of Genic and Regulatory Copy Number Variations in Bipolar Disorder, Schizophrenia, and Autism Spectrum Disorder. Biol. Psychiatry 2022, 92, 362–374.
- Kushima, I.; Aleksic, B.; Nakatochi, M.; Shimamura, T.; Okada, T.; Uno, Y.; Morikawa, M.; Ishizuka, K.; Shiino, T.; Kimura, H.; et al. Comparative Analyses of Copy-Number Variation in Autism Spectrum Disorder and Schizophrenia Reveal Etiological Overlap and Biological Insights. Cell Rep. 2018, 24, 2838–2856.
- Kushima, I.; Aleksic, B.; Nakatochi, M.; Shimamura, T.; Shiino, T.; Yoshimi, A.; Kimura, H.; Takasaki, Y.; Wang, C.; Xing, J.; et al. High-resolution copy number variation analysis of schizophrenia in Japan. Mol. Psychiatry 2016, 22, 430–440.
- Sarowar, T.; Grabrucker, A.M. Rho GTPases in the Amygdala—A Switch for Fears? Cells 2020, 9, 1972.
- Duman, J.G.; Blanco, F.A.; Cronkite, C.A.; Ru, Q.; Erikson, K.C.; Mulherkar, S.; Bin Saifullah, A.; Firozi, K.; Tolias, K.F. Rac-maninoff and Rho-vel: The symphony of Rho-GTPase signaling at excitatory synapses. Small GTPases 2021, 13, 14–47.
- Huang, G.-H.; Sun, Z.-L.; Li, H.-J.; Feng, D.-F. Rho GTPase-activating proteins: Regulators of Rho GTPase activity in neuronal development and CNS diseases. Mol. Cell. Neurosci. 2017, 80, 18–31.
- Ramos-Miguel, A.; Barr, A.M.; Honer, W.G. Spines, synapses, and schizophrenia. Biol. Psychiatry 2015, 78, 741–743.
- Mould, A.W.; Al-Juffali, N.; von Delft, A.; Brennan, P.E.; Tunbridge, E.M. Kalirin as a Novel Treatment Target for Cognitive Dysfunction in Schizophrenia. CNS Drugs 2021, 36, 1–16.
- Hanifa, M.; Singh, M.; Randhawa, P.K.; Jaggi, A.S.; Bali, A. A focus on Rho/ROCK signaling pathway: An emerging therapeutic target in depression. Eur. J. Pharmacol. 2023, 946, 175648.
- Van Bokhoven, H. Genetic and epigenetic networks in intellectual disabilities. Annu. Rev. Genet. 2011, 45, 81–104.
- Liaci, C.; Camera, M.; Caslini, G.; Rando, S.; Contino, S.; Romano, V.; Merlo, G.R. Neuronal Cytoskeleton in Intellectual Disability: From Systems Biology and Modeling to Therapeutic Opportunities. Int. J. Mol. Sci. 2021, 22, 6167.
- Guo, D.; Yang, X.; Shi, L. Rho GTPase Regulators and Effectors in Autism Spectrum Disorders: Animal Models and Insights for Therapeutics. Cells 2020, 9, 835.
- Mosaddeghzadeh, N.; Ahmadian, M.R. The RHO Family GTPases: Mechanisms of Regulation and Signaling. Cells 2021, 10, 1831.
- Symons, M.; Settleman, J. Rho family GTPases: More than simple switches. Trends Cell Biol. 2000, 10, 415–419.
- Cherfils, J.; Zeghouf, M. Regulation of small GTPases by GEFs, GAPs, and GDIs. Physiol. Rev. 2013, 93, 269–309.
- Jaiswal, M.; Dvorsky, R.; Ahmadian, M.R. Deciphering the molecular and functional basis of Dbl family proteins: A novel systematic approach toward classification of selective activation of the Rho family proteins. J. Biol. Chem. 2013, 288, 4486–4500.
- Kreider-Letterman, G.; Carr, N.M.; Garcia-Mata, R. Fixing the GAP: The role of RhoGAPs in cancer. Eur. J. Cell Biol. 2022, 101, 151209.
- Bustelo, X.R.; Sauzeau, V.; Berenjeno, I.M. GTP-binding proteins of the Rho/Rac family: Regulation, effectors and functions in vivo. BioEssays 2007, 29, 356–370.
- Amin, E.; Dubey, B.N.; Zhang, S.-C.; Gremer, L.; Dvorsky, R.; Moll, J.M.; Taha, M.S.; Nagel-Steger, L.; Piekorz, R.P.; Somlyo, A.V.; et al. Rho-kinase: Regulation, (dys)function, and inhibition. Biol. Chem. 2013, 394, 1399–1410.
- Amano, M.; Kanazawa, Y.; Kozawa, K.; Kaibuchi, K. Identification of the Kinase-Substrate Recognition Interface between MYPT1 and Rho-Kinase. Biomolecules 2022, 12, 159.
- Grassie, M.E.; Moffat, L.D.; Walsh, M.P.; MacDonald, J.A. The myosin phosphatase targeting protein (MYPT) family: A regulated mechanism for achieving substrate specificity of the catalytic subunit of protein phosphatase type 1delta. Arch. Biochem. Biophys. 2011, 510, 147–159.
- Seccia, T.M.; Rigato, M.; Ravarotto, V.; Calò, L.A. ROCK (RhoA/Rho Kinase) in Cardiovascular–Renal Pathophysiology: A Review of New Advancements. J. Clin. Med. 2020, 9, 1328.
- Luo, L. RHO GTPASES in neuronal morphogenesis. Nat. Rev. Neurosci. 2000, 1, 173–180.
- Civiero, L.; Greggio, E. PAKs in the brain: Function and dysfunction. Biochim. Biophys. Acta (BBA)—Mol. Basis Dis. 2018, 1864, 444–453.
- Dickson, B.J. Rho GTPases in growth cone guidance. Curr. Opin. Neurobiol. 2001, 11, 103–110.
- Burridge, K.; Guilluy, C. Focal adhesions, stress fibers and mechanical tension. Exp. Cell Res. 2015, 343, 14–20.
- Benarroch, E.E. Rho GTPases: Role in dendrite and axonal growth, mental retardation, and axonal regeneration. Neurology 2007, 68, 1315–1318.
- Newey, S.E.; Velamoor, V.; Govek, E.-E.; Van Aelst, L. Rho GTPases, dendritic structure, and mental retardation. J. Neurobiol. 2005, 64, 58–74.
- Sekiguchi, M.; Sobue, A.; Kushima, I.; Wang, C.; Arioka, Y.; Kato, H.; Kodama, A.; Kubo, H.; Ito, N.; Sawahata, M.; et al. ARHGAP10, which encodes Rho GTPase-activating protein 10, is a novel gene for schizophrenia risk. Transl. Psychiatry 2020, 10, 247.
- Guo, W.; Cai, Y.; Zhang, H.; Yang, Y.; Yang, G.; Wang, X.; Zhao, J.; Lin, J.; Zhu, J.; Li, W.; et al. Association of ARHGAP18 polymorphisms with schizophrenia in the Chinese-Han population. PLoS ONE 2017, 12, e0175209.
- Potkin, S.G.; Turner, J.A.; Fallon, J.A.; Lakatos, A.; Keator, D.B.; Guffanti, G.; Macciardi, F. Gene discovery through imaging genetics: Identification of two novel genes associated with schizophrenia. Mol. Psychiatry 2008, 14, 416–428.
- Potkin, S.G.; Macciardi, F.; Guffanti, G.; Fallon, J.H.; Wang, Q.; Turner, J.A.; Lakatos, A.; Miles, M.F.; Lander, A.; Vawter, M.P.; et al. Identifying gene regulatory networks in schizophrenia. NeuroImage 2010, 53, 839–847.
- Ohi, K.; Hashimoto, R.; Nakazawa, T.; Okada, T.; Yasuda, Y.; Yamamori, H.; Fukumoto, M.; Umeda-Yano, S.; Iwase, M.; Kazui, H.; et al. The p250GAP gene is associated with risk for schizophrenia and schizotypal personality traits. PLoS ONE 2012, 7, e35696.
- Gandal, M.J.; Zhang, P.; Hadjimichael, E.; Walker, R.L.; Chen, C.; Liu, S.; Won, H.; Van Bakel, H.; Varghese, M.; Wang, Y.; et al. Transcriptome-wide isoform-level dysregulation in ASD, schizophrenia, and bipolar disorder. Science 2018, 362, eaat8127.
- Kushima, I.; Nakamura, Y.; Aleksic, B.; Ikeda, M.; Ito, Y.; Shiino, T.; Okochi, T.; Fukuo, Y.; Ujike, H.; Suzuki, M.; et al. Resequencing and association analysis of the KALRN and EPHB1 genes and their contribution to schizophrenia susceptibility. Schizophr. Bull. 2010, 38, 552–560.
- Russell, T.A.; Grubisha, M.J.; Remmers, C.L.; Kang, S.K.; Forrest, M.P.; Smith, K.R.; Kopeikina, K.J.; Gao, R.; Sweet, R.A.; Penzes, P. A Schizophrenia-Linked KALRN Coding Variant Alters Neuron Morphology, Protein Function, and Transcript Stability. Biol. Psychiatry 2018, 83, 499–508.
- Gulsuner, S.; Stein, D.J.; Susser, E.S.; Sibeko, G.; Pretorius, A.; Walsh, T.; Majara, L.; Mndini, M.M.; Mqulwana, S.G.; Ntola, O.A.; et al. Genetics of schizophrenia in the South African Xhosa. Science 2020, 367, 569–573.
- Mizuki, Y.; Takaki, M.; Okahisa, Y.; Sakamoto, S.; Kodama, M.; Ujike, H.; Uchitomi, Y. Human Rho guanine nucleotide exchange factor 11 gene is associated with schizophrenia in a Japanese population. Hum. Psychopharmacol. Clin. Exp. 2014, 29, 552–558.
- Kimura, H.; Fujita, Y.; Kawabata, T.; Ishizuka, K.; Wang, C.; Iwayama, Y.; Okahisa, Y.; Kushima, I.; Morikawa, M.; Uno, Y.; et al. A novel rare variant R292H in RTN4R affects growth cone formation and possibly contributes to schizophrenia susceptibility. Transl. Psychiatry 2017, 7, e1214.
- Hsu, R.; Woodroffe, A.; Lai, W.-S.; Cook, M.N.; Mukai, J.; Dunning, J.P.; Swanson, D.J.; Roos, J.L.; Abecasis, G.R.; Karayiorgou, M.; et al. Nogo Receptor 1 (RTN4R) as a candidate gene for schizophrenia: Analysis using human and mouse genetic approaches. PLoS ONE 2007, 2, e1234.
- Sinibaldi, L.; De Luca, A.; Bellacchio, E.; Conti, E.; Pasini, A.; Paloscia, C.; Spalletta, G.; Caltagirone, C.; Pizzuti, A.; Dallapiccola, B. Mutations of the Nogo-66 receptor (RTN4R) gene in schizophrenia. Hum. Mutat. 2004, 24, 534–535.
- McCarthy, S.E.; Makarov, V.; Addington, A.M.; McClellan, J.; Yoon, S.; Perkins, D.O.; Dickel, D.E.; Kusenda, M.; Krastoshevsky, O.; Krause, V.; et al. Microduplications of 16p11.2 are associated with schizophrenia. Nat. Genet. 2009, 41, 1223–1227.
- Golzio, C.; Willer, J.; Talkowski, M.E.; Oh, E.C.; Taniguchi, Y.; Jacquemont, S.; Reymond, A.; Sun, M.; Sawa, A.; Gusella, J.F.; et al. KCTD13 is a major driver of mirrored neuroanatomical phenotypes of the 16p11.2 copy number variant. Nature 2012, 485, 363–367.
- Shibata, H.; Oishi, K.; Yamagiwa, A.; Matsumoto, M.; Mukai, H.; Ono, Y. PKNbeta interacts with the SH3 domains of Graf and a novel Graf related protein, Graf2, which are GTPase activating proteins for Rho family. J. Biochem. 2001, 130, 23–31.
- Hada, K.; Wulaer, B.; Nagai, T.; Itoh, N.; Sawahata, M.; Sobue, A.; Mizoguchi, H.; Mori, D.; Kushima, I.; Nabeshima, T.; et al. Mice carrying a schizophrenia-associated mutation of the Arhgap10 gene are vulnerable to the effects of methamphetamine treatment on cognitive function: Association with morphological abnormalities in striatal neurons. Mol. Brain 2021, 14, 21.
- Maeda, M.; Hasegawa, H.; Hyodo, T.; Ito, S.; Asano, E.; Yuang, H.; Funasaka, K.; Shimokata, K.; Hasegawa, Y.; Hamaguchi, M.; et al. ARHGAP18, a GTPase-activating protein for RhoA, controls cell shape, spreading, and motility. Mol. Biol. Cell 2011, 22, 3840–3852.
- Thompson, W.R.; Yen, S.S.; Uzer, G.; Xie, Z.; Sen, B.; Styner, M.; Burridge, K.; Rubin, J. LARG GEF and ARHGAP18 orchestrate RhoA activity to control mesenchymal stem cell lineage. Bone 2017, 107, 172–180.
- Humphries, B.; Wang, Z.; Li, Y.; Jhan, J.-R.; Jiang, Y.; Yang, C. ARHGAP18 Downregulation by miR-200b Suppresses Metastasis of Triple-Negative Breast Cancer by Enhancing Activation of RhoA. Cancer Res. 2017, 77, 4051–4064.
- Nakazawa, T.; Watabe, A.M.; Tezuka, T.; Yoshida, Y.; Yokoyama, K.; Umemori, H.; Inoue, A.; Okabe, S.; Manabe, T.; Yamamoto, T. p250GAP, a novel brain-enriched GTPase-activating protein for Rho family GTPases, is involved in the N-Methyl-d-aspartate receptor signaling. Mol. Biol. Cell 2003, 14, 2921–2934.
- Nakazawa, T.; Kuriu, T.; Tezuka, T.; Umemori, H.; Okabe, S.; Yamamoto, T. Regulation of dendritic spine morphology by an NMDA receptor-associated Rho GTPase-activating protein, p250GAP. J. Neurochem. 2008, 105, 1384–1393.
- Kannan, M.; Lee, S.-J.; Schwedhelm-Domeyer, N.; Nakazawa, T.; Stegmüller, J. p250GAP is a novel player in the Cdh1-APC/Smurf1 pathway of axon growth regulation. PLoS ONE 2012, 7, e50735.
- Paskus, J.D.; Herring, B.E.; Roche, K.W. Kalirin and Trio: RhoGEFs in Synaptic Transmission, Plasticity, and Complex Brain Disorders. Trends Neurosci. 2020, 43, 505–518.
- Penzes, P.; Johnson, R.C.; Kambampati, V.; Mains, R.E.; Eipper, B.A. Distinct roles for the two Rho GDP/GTP exchange factor domains of kalirin in regulation of neurite growth and neuronal morphology. J. Neurosci. 2001, 21, 8426–8434.
- Yan, Y.; Eipper, B.A.; Mains, R.E. Kalirin-9 and Kalirin-12 Play Essential Roles in Dendritic Outgrowth and Branching. Cereb. Cortex 2014, 25, 3487–3501.
- Deo, A.J.; Cahill, M.E.; Li, S.; Goldszer, I.; Henteleff, R.; VanLeeuwen, J.-E.; Rafalovich, I.; Gao, R.; Stachowski, E.K.; Sampson, A.R.; et al. Increased expression of Kalirin-9 in the auditory cortex of schizophrenia subjects: Its role in dendritic pathology. Neurobiol. Dis. 2012, 45, 796–803.
- Grubisha, M.J.; Sun, T.; Eisenman, L.; Erickson, S.L.; Chou, S.-Y.; Helmer, C.D.; Trudgen, M.T.; Ding, Y.; Homanics, G.E.; Penzes, P.; et al. A Kalirin missense mutation enhances dendritic RhoA signaling and leads to regression of cortical dendritic arbors across development. Proc. Natl. Acad. Sci. USA 2021, 118, e2022546118.
- Harrington, A.W.; Li, Q.M.; Tep, C.; Park, J.B.; He, Z.; Yoon, S.O. The role of Kalirin9 in p75/nogo receptor-mediated RhoA activation in cerebellar granule neurons. J. Biol. Chem. 2008, 283, 24690–24697.
- Yamashita, T.; Tucker, K.L.; Barde, Y.-A. Neurotrophin binding to the p75 receptor modulates Rho activity and axonal outgrowth. Neuron 1999, 24, 585–593.
- Boghdadi, A.G.; Teo, L.; Bourne, J.A. The Involvement of the Myelin-Associated Inhibitors and Their Receptors in CNS Plasticity and Injury. Mol. Neurobiol. 2017, 55, 1831–1846.
- Rümenapp, U.; Blomquist, A.; Schwörer, G.; Schablowski, H.; Psoma, A.; Jakobs, K.H. Rho-specific binding and guanine nucleotide exchange catalysis by KIAA0380, a dbl family member. FEBS Lett. 1999, 459, 313–318.
- Fukuhara, S.; Murga, C.; Zohar, M.; Igishi, T.; Gutkind, J.S. A Novel PDZ domain containing guanine nucleotide exchange factor links heterotrimeric G proteins to Rho. J. Biol. Chem. 1999, 274, 5868–5879.
- Jackson, M.; Song, W.; Liu, M.-Y.; Jin, L.; Dykes-Hoberg, M.; Lin, C.-L.G.; Bowers, W.J.; Federoff, H.J.; Sternweis, P.C.; Rothstein, J.D. Modulation of the neuronal glutamate transporter EAAT4 by two interacting proteins. Nature 2001, 410, 89–93.
- Mizuki, Y.; Takaki, M.; Sakamoto, S.; Okamoto, S.; Kishimoto, M.; Okahisa, Y.; Itoh, M.; Yamada, N. Human Rho Guanine Nucleotide Exchange Factor 11 (ARHGEF11) Regulates Dendritic Morphogenesis. Int. J. Mol. Sci. 2016, 18, 67.
- Mizuki, Y.; Sakamoto, S.; Okahisa, Y.; Yada, Y.; Hashimoto, N.; Takaki, M.; Yamada, N. Mechanisms Underlying the Comorbidity of Schizophrenia and Type 2 Diabetes Mellitus. Int. J. Neuropsychopharmacol. 2020, 24, 367–382.
- Davidkova, G.; Mccullumsmith, R.E.; Meador-Woodruff, J.H. Expression of ARHGEF11 mRNA in schizophrenic thalamus. Ann. N. Y. Acad. Sci. 2003, 1003, 375–377.
- Karayiorgou, M.; Simon, T.J.; Gogos, J.A. 22q11.2 microdeletions: Linking DNA structural variation to brain dysfunction and schizophrenia. Nat. Rev. Neurosci. 2010, 11, 402–416.
- Schwab, M.E. Functions of Nogo proteins and their receptors in the nervous system. Nat. Rev. Neurosci. 2010, 11, 799–811.
- Perlstein, M.D.; Chohan, M.R.; Coman, I.L.; Antshel, K.M.; Fremont, W.P.; Gnirke, M.H.; Kikinis, Z.; Middleton, F.A.; Radoeva, P.D.; Shenton, M.E.; et al. White matter abnormalities in 22q11.2 deletion syndrome: Preliminary associations with the Nogo-66 receptor gene and symptoms of psychosis. Schizophr. Res. 2014, 152, 117–123.
- Fernandez-Enright, F.; Andrews, J.L.; Newell, K.A.; Pantelis, C.; Huang, X.F. Novel implications of Lingo-1 and its signaling partners in schizophrenia. Transl. Psychiatry 2014, 4, e348.
- Jitoku, D.; Hattori, E.; Iwayama, Y.; Yamada, K.; Toyota, T.; Kikuchi, M.; Maekawa, M.; Nishikawa, T.; Yoshikawa, T. Association study of Nogo-related genes with schizophrenia in a Japanese case-control sample. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2011, 156, 581–592.
- Lin, G.N.; Corominas, R.; Lemmens, I.; Yang, X.; Tavernier, J.; Hill, D.E.; Vidal, M.; Sebat, J.; Iakoucheva, L.M. Spatiotemporal 16p11.2 protein network implicates cortical late mid-fetal brain development and KCTD13-Cul3-RhoA pathway in psychiatric diseases. Neuron 2015, 85, 742–754.
- Chen, Y.; Yang, Z.; Meng, M.; Zhao, Y.; Dong, N.; Yan, H.; Liu, L.; Ding, M.; Peng, H.B.; Shao, F. Cullin mediates degradation of RhoA through evolutionarily conserved BTB adaptors to control actin cytoskeleton structure and cell movement. Mol. Cell 2009, 35, 841–855.
- Genschik, P.; Sumara, I.; Lechner, E. The emerging family of CULLIN3-RING ubiquitin ligases (CRL3s): Cellular functions and disease implications. EMBO J. 2013, 32, 2307–2320.
- Urresti, J.; Zhang, P.; Moran-Losada, P.; Yu, N.-K.; Negraes, P.D.; Trujillo, C.A.; Antaki, D.; Amar, M.; Chau, K.; Pramod, A.B.; et al. Cortical organoids model early brain development disrupted by 16p11.2 copy number variants in autism. Mol. Psychiatry 2021, 26, 7560–7580.
More