Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Gianluigi Mazzoccoli and Version 2 by Rita Xu.
Neuronal PAS domain protein 2 (NPAS2) is a hemeprotein comprising a basic helix–loop–helix domain (bHLH) and two heme-binding sites, the PAS-A and PAS-B domains.
- NPAS2
- circadian
- rhythmicity
- molecular clockwork
1. Introduction
NPAS2 (in extensum Neuronal-PAS type signal-sensor protein-domain protein 2), alias member of PAS protein 4 (MOP4), is a protein coding gene, in mammals largely expressed in the forebrain. In Mus musculus the Npas2 gene is 169.505 bases long and is located to chromosome 1 B; 1 at approximately 2 centimorgans, whereas in Homo sapiens NPAS2 is 176.679 bases long, contains 25 exons and is located to chromosome 2q11.2, close to the centromere [1][2][1,2]. Human NPAS2 protein contains 824 amino acids with a molecular mass of 91791 Da and has in common 87% of the sequence with mouse Npas2 protein (816 amino acids), indicating that the mouse and human genes are true homologs [1][2][1,2] (Figure 1). NPAS2 represents a main component of the circadian molecular clock, with distinguishing structural and functional features that allow the binding of heme and act as gas-responsive transcription factor [3]. On the basis of these features, NPAS2 is particularly important for circadian transcriptional events and intracellular signaling, especially in mammalian brain structures and neural circuits. To address the role of NPAS2 in physiological processes, such as: (i) the functioning of the circadian clock circuitry, (ii) the regulation of metabolic pathways, (iii) the function of the mammalian central nervous and cardiovascular systems, and (iv) wound healing. Furthermore, NPAS2 is implicated in pathological mechanisms of disease, such as those pushing cancer onset and progression.
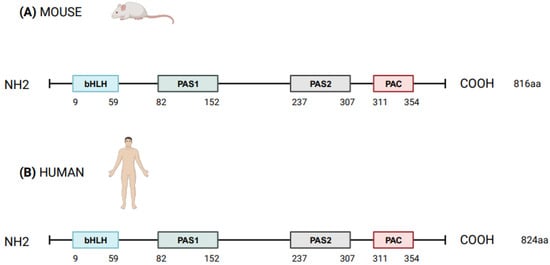
Figure 1. Schematic structure of NPAS2 protein in Mus Musculus (A) and Homo sapiens (B). bHLH = basic helix–loop–helix; PAS = Period–Arnt–Single-minded; PAC = PAS-associated C-terminal.
2. The Circadian Timing System
Fundamental cellular processes as well as tissue and organ functions are characterized by rhythmic fluctuations with prevalent 24 h periodicity, defined circadian timing and are driven by molecular clockworks endowing every single cell in the body [4]. These cell-autonomous and self-sustained peripheral oscillators are synchronized by leading oscillators in the central nervous system and organized at tissue, organ and organ system level to arrange a hierarchical network structure-defined circadian timing system [5][6][7][8][5,6,7,8]. In particular, a master pacemaker is located in the suprachiasmatic nuclei (SCNs) of the hypothalamus. SCNs synchronize rhythmic activity in other brain regions and centers, through direct monosynaptic projections, and in peripheral tissues, through autonomic nervous system fibers and neuroendocrine outputs (primarily cortisol and melatonin) [9][10][11][9,10,11] (Figure 2). SCNs are located in the anterior part of the hypothalamus, on each side of the third ventricle, in the area situated directly above the optic chiasm and consist of roughly 8000–10,000 neurons in Mus musculus and 80.000–100.000 neurons in Homo sapiens [12]. SCNs receive photic inputs through the retino-hypothalamic tract (RHT), which conveys signals from intrinsically photosensitive retinal ganglion cells (ipRGCs), a subpopulation representing less than 5% of retinal ganglion cells [13][14][13,14]. ipRGCs signal light stimuli synchronizing and entraining physiological circadian rhythmicity to the foremost environmental cue, represented by light/darkness alternation linked to the daily rotation of planet Earth on its axis (photo-entrainment) [15] (Figure 2). In the SCN it is possible to identify “core” and “shell” sub-regions expressing different neuropeptides, crucial for circadian rhythmicity maintenance: vasoactive intestinal peptide (VIP) and gastrin-releasing peptide (GRP) are mainly expressed in the retino-recipient core, whereas arginine vasopressin (AVP) is expressed in the shell [16]. The activity of the core and shell VIP-, GRP-, and AVP-expressing neurons is stringently ordered in a time-qualified manner. VIP and VPAC2 receptors increase in the core sub-region during nighttime and uphold SCN internal synchronization. GRP is definitely stimulated by direct inputs from ipRGCs via the RHT, increases during the morning and peaks around midday [17][18][19][20][17,18,19,20]. AVP-expressing neurons, prevalently located in the “shell” sub-region, project to the paraventricular nucleus (PVN), which coordinates feeding–fasting with sleep–wake and rest–activity cycles, and to thirst-controlling neurons in the organum vasculosum lamina terminalis (OVLT) [19][20][19,20]. In the hypothalamic SCNs, circadian fluctuations of gene expression and electrical activity can continue relentlessly in isolated conditions (no photic cues). This robust and resilient time-keeping is produced inherently and is largely related to the intrinsic connectivity of SCN neurons [21][22][21,22]. Indeed, SCN circuit-level time-keeping is operated through mutually dependent astrocytic–neuronal signaling. SCN neurons are metabolically active during daytime, whereas SCN astrocytes are active during nighttime and drive fluctuations of glutamate levels in the extracellular space, peaking in circadian night. Neurons of the dorsal SCN gauge this glutamatergic glio-transmission via specific presynaptic NMDA receptor assemblies containing NR2C subunits [21][22][21,22]. Upon glutamate binding through NR2C subunit-containing NMDA-type glutamate receptors, intracellular Ca2+ concentration increases in presynaptic neurons. They release gamma-aminobutyric acid (GABA) that suppresses the electrical activity of postsynaptic neurons [21][22][21,22]. Subsequently, extracellular glutamate is transported back in astrocytes by excitatory amino acid transporters, and presynaptic GABAergic tone decreases and electrical firing of postsynaptic neurons increases. In this way, oscillations of intracellular Ca2+, extracellular glutamate and GABAergic signaling maintain astrocytic–neuronal interplay in the SCNs [21][22][21,22].
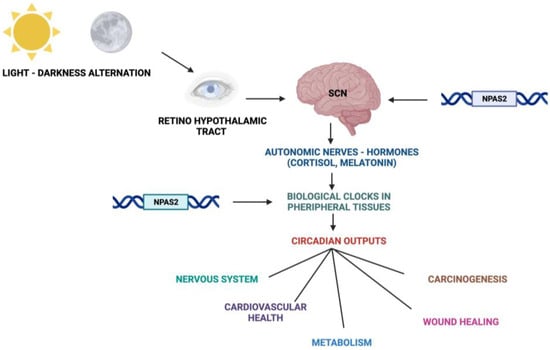
Figure 2. Schematic representation of the Circadian Timing System with functions and processes in which NPAS2 is involved.
3. NPAS2 and the Molecular Clockwork
At the cellular level, the cogs that run the molecular clockwork driving rhythmicity of cell functions and processes are embodied by a group of circadian proteins encoded by genes, also known as (core) clock genes, operating interlocking transcription–translation feedback loops (TTFL). TTFL are organized into an activator arm and an inhibitory arm that interact mutually in a time frame including a delay that determines the completion of a regular cycle of interaction in approximately 24 h [23]. In detail, the bHLH-PAS (basic helix–loop–helix–Period–Arnt–Single-minded) transcriptional activators CLOCK (circadian locomotor output cycles kaput), and its paralog NPAS2 (neuronal PAS domain protein 2), and ARNTL-2/BMAL1-2 (aryl-hydrocarbon receptor nuclear translocator-like/brain and muscle aryl-hydrocarbon receptor nuclear translocator-like) operate the positive limb of the TTFL, heterodimerizing and binding to enhancer (E)-box DNA consensus sequences of the target genes Cryptochrome (CRY 1-2) and Period (PER1-3), which operate the negative limb of the TTFL [24]. NPAS2 can compensate for the loss of the core transcription factor and histone/protein acetylase CLOCK in the molecular clockwork endowing SCN oscillators as well as peripheral tissues cells [25][26][27][25,26,27]. The circadian proteins CRY1-2 and PER1-3 accumulate in the cytoplasm and dimerize establishing repressor complexes that pass back into the nucleus and hinder the transcriptional activity of ARNTL:NPAS2 and ARNTL:CLOCK heterodimers [28][29][30][31][28,29,30,31]. The circadian proteins go through post-translational modifications (PTMs), such as phosphorylation, acetylation, sumoylation, O-GlcNAcylation, which allow activity modulation and sequential ubiquitination/proteasomal degradation: this is crucial for appropriate functioning of the circadian clock circuitry [32][33][34][32,33,34]. The nuclear receptors (NRs) REV-ERBs and retinoic acid-related (RAR) orphan receptor (RORs) manage an auxiliary interconnected loop driving ARNTL rhythmic transcription competing with ROR specific response elements (RORE) in its promoter [35]. ROR-α physically interacts with peroxisome proliferator-activated receptor (PPAR)-γ coactivator-1α (PGC-1α) and acts as transcriptional activator and recruits chromatin-remodeling complexes to proximal gene promoters and prompts ARNTL transcription. Contrariwise, REV-ERBs inhibits ARNTL transcription interacting with the nuclear corepressor/histone deacetylase3 (NCoR-HDAC3) corepressor complex [35].
Circadian transcription of the NPAS2 gene, likewise ARNTL transcription, is regulated and synchronized through RORs and REV-ERBs competition at ROR and REV-ERB response elements (ROREs and Rev-REs, respectively) in the upstream region of the transcription start site [36][37][36,37]. Additionally, a specific activating role is played by RORγ in the regulation of NPAS2 expression through direct binding onto two ROREs in its proximal promoter [38].
In addition to the NR-operated feedback loop, ARNTL:NPAS2 and ARNTL:CLOCK heterodimers bind to cognate D-box elements and drive the expression of first order clock controlled genes, which encode the PAR domain basic leucine zipper (bZIP) transcription factors DBP (D site of albumin promoter—Albumin D-box—binding protein), TEF (thyrotroph embryonic factor), and HLF (hepatic leukaemia factor). DBP, TEF and HLF drive the rhythmic expression of thousands of tissue specific (output) genes [39][40][39,40].
Moreover, RORs and DBP bind to distinct response elements in the promoter—the RORE and the DNA cis-elements (D-box), respectively—and drive the rhythmic expression of Nuclear factor, interleukin 3 regulated (NFIL3, also known as E4BP4) [39][40][39,40]. In order, DBP and NFIL3 protein levels and binding to D-box elements on target genes oscillate in opposite phases, driving D-box-dependent rhythmic expression of REV-ERBs, RORs and PER genes [41][42][43][44][41,42,43,44] (Figure 3).
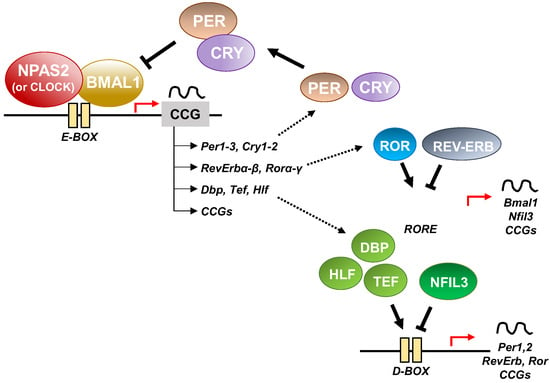
Figure 3. Schematic representation of the molecular clockwork.
Other key molecular cogs of the circadian clock circuitry are the bHLH transcription factors differentially expressed in chondrocytes protein 1 (DEC1) and 2 (DEC2), which interplay with the core clock proteins. In particular, DEC1 and DEC2 transcription is activated by ARNTL:NPAS2 and ARNTL:CLOCK heterodimers, but in sequence DEC1 and DEC2 proteins block their transcriptional activity, managing a feedback steering circuit [39][40][45][39,40,45]. The functioning of the molecular clockwork is finely tuned through rhythmic chromatin-histone remodeling and epigenetic modifications, principally geared up by acetylation/deacetylation and methylation/demethylation processes [46]. Concerning the cogs of the molecular clockwork, ARNTL is acetylated by CLOCK, which has intrinsic protein and histone acetyltransferase capability [47]. Conversely, the type III histone/protein deacetylase SIRT1 opposes this process. Its deacetylating activity relies on the intracellular levels of nicotinamide adenine dinucleotide (NAD+), a nutrient sensor produced from tryptophan through the enzymatic activity of nicotinamide phosphoribosyl-transferase (NAMPT/visfatin), which is expressed rhythmically driven by the circadian clock circuitry [48][49][50][51][48,49,50,51].
In summary, entangled TTFLs set up the essential molecular hardware driving circadian rhythmicity in mammals. Negative arms of the feedback loops are operated by CRY/PER proteins, which hinder the transcriptional action of activator circadian proteins ARNTL and CLOCK/NPAS2. On the other hand, ROR and REV-ERB nuclear receptors prompt and hamper, respectively, the expression of genes encoding the activator cogs. In particular, the role of NPAS2 in the circadian molecular clock as an obligate dimeric partner of ARNTL is important in mammals, especially in the central nervous system and in metabolically active tissues such as the liver. Alteration of the balance between these positive and negative limbs due to genomic, genetic and epigenetic modifications in the circadian proteins operating the TTFL, and NPAS2 in particular, can cause a range of pathological conditions.
4. NPAS2, Hemeprotein and Gas-Responsive Transcription Factor
A protein in which a heme-binding domain controls the activity of another domain is defined as a heme-based sensor. NPAS2 contains a heme-binding motif and heme controls the transcriptional activity of the ARNTL:NPAS2 heterodimer [52]. NPAS2 is a pyridine nucleotide-dependent and carbon monoxide (CO)-dependent transcription factor consisting of a stabilizing and DNA-binding bHLH domain, two heme-binding PAS (Period–Arnt–Single-minded) domains in the N-terminal region and one PAC (PAS-associated C-terminal) domain in the C-terminal region. In order, the presence of heme in NPAS2 is linked to heme metabolism, which is rhythmically controlled by the circadian clock circuitry, bringing on a mutual regulation. Indeed, the molecular clockwork drives the circadian expression of ALAS1, the gene coding for 5′-aminolevulinate synthase 1, the rate-limiting enzyme in the biochemical pathway of heme synthesis [53]. PAS-A and PAS-B domains of NPAS2 bind heme as a prosthetic group, realizing a gas-regulated sensor that modifies DNA binding in vitro depending on heme status.
Well-designed resonance Raman (RR) experiments coupled with mutational studies performed on a mouse isolated PAS-A domain [54] shed light on the iron coordination sphere of the ferric, ferrous and carbonylated states of the protein and on the spin state of the heme. The overall conclusion of this work is that both the ferric and ferrous PAS-A domains consist of a mixture of five- and six-coordinate heme. The resonance Raman spectra of the isolated PAS-A domain have been thoroughly assessed in various studies [54][55][56][54,55,56]. When excited at 363.8 nm, a spectral band related to the stretching of Fe3+-S- was observed at 334 cm−1 in the ferric protein, where Cys170 was identified as an axial ligand for the ferric heme [54][55][54,55]. The Raman spectrum of the reduced form mostly exhibited a six-coordinate low-spin configuration. Notably, the n11 band, sensitive to the donor strength of the axial ligand, appeared lower compared to reduced cytochrome c3, suggesting the presence of a strong ligand, likely a deprotonated His [54][55][54,55]. In the reduced forms of H119A and H171A mutants, five-coordinate species were more prevalent, while no such alterations were observed for C170A, indicating that His119 and His171, but not Cys170, serve as axial ligands in the ferrous heme [54][55][54,55]. These findings suggest that there is a transition from Cys to His as the axial ligand upon heme reduction, and the nFe-CO versus nC-O correlation indicates that a neutral His acts as a trans ligand to CO [54][55][54,55]. In the isolated PAS-A domain, it was determined that His119 and Cys170 serve as axial ligands for the ferric heme. However, upon heme reduction, Cys170 is replaced by His171. The coordination structure of the isolated PAS-A domain exists in an equilibrium between Cys–Fe–His and His–Fe–His coordinated species, but when interacting with the bHLH domain, the equilibrium shifts towards the latter configuration [54][55][54,55]. The fragment containing the N-terminal bHLH of the first PAS (PAS-A) domain of NPAS2 predominantly forms dimers in solution. The Soret absorption peak of the ferric complex for bHLH-PAS-A (421 nm) exhibits a 9 nm red shift compared to isolated PAS-A (412 nm). Based on RR spectra, it appears that His is the axial ligand trans to CO in bHLH-PAS-A. Furthermore, the rate constant for heme association with apo-bHLH-PAS is more than two orders of magnitude higher than that for association with apo-PAS-A [56]. Optical absorption spectra of the PAS-B domain (residues 241–416) of mouse NPAS2 revealed that Fe(III), Fe(II), and Fe(II)-CO complexes are six-coordinate low-spin complexes. In contrast, resonance Raman spectra indicated that both Fe(III) and Fe(II) complexes contain mixtures of five-coordinate high-spin and six-coordinate low-spin complexes [57].
Overall, the emerging picture of the heme environment is complex and most likely dynamic. Reduction of heme iron (by indefinite electron donors) results in an endogenous ligand exchange reaction whereby Cys170 is exchanged for His171 in the ferrous state. To bind exogenous ligands, one of the His ligands must dissociate from the ferrous ion (Figure 4).
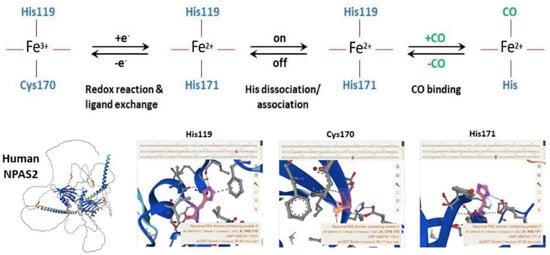
Figure 4. Molecular structure and biochemistry of the circadian hemeprotein and gas-responsive transcription factor NPAS2. TOP Redox, endogenous ligand dissociation/association and CO binding scheme of the PAS-A domain. BOTTOM Structure images of NPAS2 with His119, Cys170 and His171 exchange.
The apo (heme-free) or holo (heme-loaded) states of ARNTL:NPAS2 heterodimers strongly bind DNA under favorably reducing ratios of NADPH/NADP+, while micromolar concentrations of CO impede the DNA-binding activity of holo-NPAS2, but not of apo-NPAS2 [58][59][58,59]. Precisely, CO concentrations above 3 µM are capable of hindering the binding of holo-NPAS2 to DNA [58][59][58,59]. Unproductive ARNTL-ARNTL homodimers form if either or both conditions that inhibit formation of the ARNTL:NPAS2 heterodimer are present [58][59][58,59]. The transcriptional activity of ARNTL:NPAS2 and ARNTL:CLOCK heterodimers is regulated by the redox state of NAD+ cofactors. NAD(H) and NADP(H), the reduced forms of the redox cofactors, powerfully enhance DNA binding of ARNTL:NPAS2 and ARNTL:CLOCK heterodimers, while NAD+ and NADP+, the oxidized forms, hinder DNA-binding capability [58]. Mouse NPAS2 and ARNTL truncated forms, containing only the corresponding bHLH domains and no PAS domain, completely retained NADPH-dependent DNA-binding capability [58]. These data suggest that: (i) the NAPDH binding site must reside on the bHLH domain; (ii) NADPH does not undergo redox changes in the bound state and behaves as an allosteric ligand; (iii) alternatively, in situ NAPDH auto-oxidation could fix the lifetime of the DNA-heterodimer complex [59]. A significant portion of the cellular amount of NADPH derives from the pentose phosphate pathway, whose activity oscillates with circadian rhythmicity driven by the molecular clockwork through the redox-sensitive transcription factor NRF2 [60]. In this way, the NADP+/NADPH ratio, with NADPH serving as an electron donor, mediates the rhythmic interplay between the cellular redox state and circadian transcriptional events [60]. Along with the enhancing activity of NADPH levels, changes of cell pH from 7.0 to 7.5 increases the DNA-binding capability of NPAS2, whose N-terminal amino acids 1–61 result necessary to sense the change in both pH and NADPH levels [61].
Above and beyond heme binding, NPAS2 can reversibly bind CO in vitro and in vivo. CO binds the heme group in NPAS2 and inhibits the DNA-binding activity of the ARNTL:NPAS2 heterodimer. Endogenous CO is continuously produced during heme metabolism and modulates the DNA-binding patterns of NPAS2 and CLOCK onto the promoters of circadian genes [62]. Remarkably, heme degradation is regulated by the molecular clockwork: transcript and activity levels of Heme oxygenase (Ho)-1, the main heme-degrading enzyme, fluctuate with circadian rhythmicity. In turn, cyclical degradation of heme controlled by HO-1 determines the rhythmic increase in heme-degradation products, such as biliverdin, iron and CO [63].
Experiments performed in a mouse model treated with hemoCD1 (a supramolecular complex of iron(II)porphyrin with a per-O-methyl-β-cyclodextrin dimer and with highly selective CO scavenging action) showed that endogenous CO removal leads to up-regulation of the E-box-controlled circadian genes Per1, Per2, Cry1, Cry2, and Rev-erbα in the liver, corroborating the role played by endogenous CO in the regulation of the mammalian circadian clock [62][63][64][65][66][62,63,64,65,66].
Overall, these data indicate that in mammals NPAS2 binds heme with both PAS domains. Binding of these prosthetic groups modifies the in vitro DNA affinity of NPAS2 upon obligate heterodimerization with ARNTL. High reduced/oxidized NAD+ and NADP+ ratios increase the DNA binding of holo (heme-loaded) and apo (heme-free) NPAS2. Furthermore, DNA binding of holo-NPAS2, but not apo-NPAS2, is inhibited by micromolar concentrations of CO. Collectively, these data suggest that gaseous signaling in addition to heme-based sensing modulates the transcriptional activity of NPAS2 and the expression of its target genes, thereby impacting the important signaling pathways that they enrich.
5. NPAS2 and the Metabolic Pathways
Core circadian genes and tissue-specific output genes drive rhythmic expression of hundreds of transcripts enriching metabolic pathways involved in glucose and lipid metabolism as well as xenobiotic detoxification. Likewise, deregulation of circadian genes plays a crucial role in the pathophysiological mechanisms underlying metabolic derangements [67]. In the liver, Npas2 with the nuclear receptor and transcriptional regulator small heterodimer partner (SHP) realizes a feedback regulatory loop involved in triglyceride and lipoprotein homeostasis. SHP is a bona fide transcriptional repressor of Npas2 and acts at the molecular level as a negative controller of nuclear receptor-dependent signaling pathways, suppressing Rorγ transactivation and acting as a co-repressor of Rev-erbα. SHP enhances Rev-erbα inhibitory action on the positive transcriptional activity of Rorα at the Npas2 promoter, with subsequent inhibition of Npas2 transcription [68]. On the contrary, Npas2 binds to the Shp promoter and drives circadian Shp gene expression. In its turn SHP, interplaying with RORα, RORγ, or REV-ERBα, modulates the regulatory role played by Npas2 in upholding 24 h rhythms of lipid metabolism. As a result, Shp−/− mice are characterized by the severe derangement of time-qualified patterns of expression of fundamental genes implicated in cholesterol, fatty acid, bile acid, and lipid metabolism in the liver [68]. Experiments performed in vitro in hepatic cells (AML-12, Hepa1-6 and HepG2 cells) and in vivo in Npas2−/− mice showed that NPAS2 drives 24 h rhythms of expression and activity of hepatic CYP1A2. Indeed, NPAS2 trans-activates its expression through specific binding to the -416 bp E-box-like element within the Cyp1a2 gene promoter [69]. CYP1A2 is a monooxygenase member of the cytochrome P450 enzyme superfamily that catalyzes numerous reactions involved in drug metabolism and in the synthesis of cholesterol, steroids, and other lipids. CYP1A2 also intervenes in the metabolism of polyunsaturated fatty acids, transforming them into signaling molecules involved in physiological processes and pathophysiological mechanisms of disease. Likewise, NPAS2 was found crucial in determining time-qualified patterns of toxicity of brucine, a foremost bioactive and toxic component of the herb drug Semen Strychni. Brucine hepatotoxicity was assessed through transaminase levels in plasma and analysis of liver histopathology in wild type and Npas2−/− mice. The latter showed significant down-regulation of Cyp3a11 expression, with a loss of circadian rhythmicity in brucine pharmacokinetics, liver distribution and hepatotoxicity [70].
In summary, these data indicate that NPAS2 plays an important role for the regulation of hepatic metabolism, driving the rhythmic transcription of genes encoding enzymatic proteins controlling key metabolic pathways in the liver.