Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Jessie Wu and Version 1 by Mukul Jain.
Alzheimer’s disease (AD) is a devastating neurodegenerative disorder characterized by progressive cognitive decline and memory loss. Early and accurate diagnosis of AD is crucial for implementing timely interventions and developing effective therapeutic strategies. Proteome-based biomarkers have emerged as promising tools for AD diagnosis and prognosis due to their ability to reflect disease-specific molecular alterations.
- Alzheimer’s disease
- biomarkers
- proteomics
- microarray
- bioinformatics
1. Amyloid-Beta (Aβ) Peptides and Tau Protein
Current disease models show that Amyloid-beta in either non-fibrillary, soluble, oligomer, or plaque form initiates tau misfolding and assembly through a pathophysiological cascade that helps in its spread throughout the cortex, causing neuronal system failure, neurodegeneration, and cognitive decline [90][1]. The amyloid precursor protein (APP) and Presenilin mutations cause the accumulation of pathological amyloid-β protein (Aβ) species in the brain resulting in early-onset ADlzheimer’s disease (AD) pathogenesis [91][2]. Normally, the beta and gamma-secretase enzymes generate soluble amyloid-beta fragments by cleaving the Amyloid-PPrecursor (APP) protein. However, a mutated condition in the APP gene forms insoluble Aβ fragments that eventually convert into clumps. These toxic Aβ species manipulate the normal tau phosphorylation regulating the function of protein kinases and phosphatases, inducing tau misfolding, and tangle formation [92][3]. Aβ pathogenicity requires tau toxicity as the tau mediates synaptic dysfunctioning and neuronal cell death, thereby enhancing memory deterioration and cognitive impairment in AD [93][4]. It is hypothesized that amyloid beta generation is initiated during the early stages of Alzheimer’s disease, and eventually uprises forming plaque deposits that progressively increase in size and downregulate glutamatergic transmission and damage the associated synapses [94][5]. A network dysfunction is generated at the site, close to deteriorated synapses where the microglial cells exhibit a response to remove the damaged synapses and prevent further damage. However, as the amyloid-β plaque deposition spreads across multiple regions in the brain, the synaptic damage becomes more prominent and spreads, causing tau hyperphosphorylation, tau dissociation from microtubules, and the tau entangle formation, which promotes axon loss and neurodegeneration [95][6]. These changes in normal brain activity and functioning link with memory loss, cognitive impairments, and brain connectivity dysregulation in a stage-dependent manner, suggesting they may be useful for tracking disease progression. Yan Li et al., 2022, established the discovery of probable amyloid-beta plaques from the CSFAβ42/40 ratio, suggesting their potential as a biomarker for AD detection [96][7]. Hansson et al., 2019, also focused on determining the effect of pre-analytical handling of biomarkers of AD and the quantity retrieved [97][8]. Similarly, Lih-Fen Lue et al., 2017, emphasized finding these Amyloid-beta and tau proteins in the blood. However, in clinical practice, CSF biomarker analysis involves sampling from patients with atypical or mixed presentation of dementia, making the diagnosis complex, thereby highlighting the importance of AD discrimination from other neurodegenerative processes [98][9].
2. Apolipoprotein E (APOE)
Apolipoprotein or ApoE, a 34 kDa glycoprotein with a 299 amino acid long polypeptide chain is a blood–brain barrier (BBB) impermeable protein, present in significant amounts in the central nervous system due to expression of astrocytes, microglia, vascular mural cells, choroid plexus cells [99][10]. In the CNS, ApoE plays a prominent role in axonal growth and synapse formation, which are crucial for learning, memory generation, and neuronal repair by delivering cholesterol to nerve cells. It is associated with the low-density lipoprotein receptor (LDLR), and LDLR-related protein 1 (LRP1) so as to maintain lipid homeostasis via lipid transport from one tissue or cell type to another [100][11]. Three varied APOE alleles exist in the human body, namely: the ε2 (APOE2), ε3 (APOE3), and ε4 (APOE4). They are distinctive from each other by the varying cysteine and arginine amino acids at the positions 112 and 158 (apoE2: Cys112/Cys158; apoE3: Cys112/Arg158; apoE4: Arg112/Arg158) and they contrastingly regulate the cholesterol levels for γ-secretase activity and Aβ production [101][12]. Genome-wide association studies deduced ε4 allele polymorphism of APOE as a significant genetic risk factor that deposits with Aβ in amyloid plaques causing late-onset AD [102][13]. The apoE4 hinders the LRP1 (low-density lipoprotein receptor-related protein 1) receptor-mediated Aβ clearance as it weakly associates with Aβ causing hindrance in the uptake of Aβ/ApoE complexes in neurons [103][14]. In a normalized state, neuronal apoE4 promotes tau phosphorylation and cell death by modulating microglial activation, however, impaired apoE4 dysregulates homeostatic microglial functioning playing a role in amyloid plaque degradation due to its reduced affinity to TREM2 (triggering receptor expressed on myeloid cells 2) receptors expressed by microglial cells [104][15]. Recent research demonstrated that ApoE4, independent of Aβ, elicits an inflammatory pathway causing neurovascular dysfunction, including blood–brain barrier collapse, leakage of blood-derived toxic proteins into the brain, and shortened small vessel length [105][16]. Thus, obtaining APOE genotype status has been commended as a necessity for AD therapy as it is a determining factor of AD risk exerting influence on multiple disease pathways [106][17]. Ying et al., 2021 focus on the elevated CSF ApoE association with longitudinal changes in AD biomarkers including Amyloid-beta and others [107][18]. Other work by Matthew Paul et al., 2022, focused on finding the imbalance between the different glycoforms of ApoE monomers in AD that cause hindrance with its biological function, contributing to the progression of the disease [108][19]. However, the presence of a lower amount of APOE4 in CSF, along with a limited sample volume of CSF, lowers the sensitivity of APOE4 detection assays. Thus, for future inventory purposes and drug development for AD, studies need to explore the therapeutic tools for analyzing and modifying certain parameters of ApoE, such as its structure and homeostasis maintaining property, thereby producing changes in pathological AD progression.
3. Clusterin (CLU) and Other Chaperone Proteins
Alzheimer’s disease (AD) is a persistent and distressing neurological condition affecting older-age populations more frequently. Some proteins, such as clusterin (CLU) or apolipoprotein J (APOJ), have been found to be associated with dementia, neurological inflammation, and oxidative stress during such AD conditions [109][20]. Clusterin, encoded by the CLU gene located on the p21-p12 locus on chromosome 8 of humans, is an omnipresent and obstinately produced protein renowned as a molecular chaperone expressed by a variety of tissues and body fluids [110][21]. It is the third-most important genetic risk element for late-onset AD, with a number of variants. The interaction and binding properties of clusterin with Aβ appear to influence aggregation and enhance Aβ clearance, hinting toward the neuroprotective effect [111][22]. Clusterin inhibits aggregation and helps LRP2 (megalin) in removing Aβ. Tau pathology spreads from cell to cell in a manner similar to prion disease that also may be regulated by extracellular chaperones like Clusterin [112][23]. Using CLU-deficient animals as models of amyloidosis, the relationship between CLU and Aβ may be discovered in vivo [113][24]. In contrast to controls, people with Alzheimer’s disease (AD) have higher levels of the mRNA, or messenger ribonucleic acid, of CLU in various parts of the cerebral tissue (brain), according to a study. Further, higher amounts of CLU proteins were also observed in both the hippocampus and frontal cortical regions of post-mortem AD brains [114][25]. A study using SH-SY5Y cells exposed to AD patients’ CSF shows the cytoprotective ability of Clusterin alone, and also in combination with extracellular chaperones it preserved and protected the cells from damage [115][26]. Under physiological circumstances, CLU reduces aggregates and mediates Aβ clearance. The co-culture studies on rat hippocampus astrocytes and neurons revealed that Clusterin incubation reduces Aβ-induced astrocytic calcium intake, resulting in diminished ROS formation and caspase 3 activations [116][27]. A more recent experiment using APP/PS1-mutated mice revealed that Clusterin knockout increases amyloid angiopathy while reducing bleeding and inflammation by shifting Aβ deposition from plaque to deposit in the cerebrovascular fluid. In a cellular model, Clusterin inhibited the development of Tau fibrils but promoted the formation of Tau oligomers to initiate the aggregation of endogenous Tau. Pre-aggregated Aβ was incubated with Clusterin, and this reduced the amount of amyloid that human primary astrocyte cultures and microglia ingested from preparations of fibrils and oligomers [117][28]. However, this transporter, CLU, is also operative at the blood–brain barrier (BBB), which serves as a physical barrier between the outside and inside of the brain. CLU-linked molecular pathways at the BBB’s interface are involved in the development and progression of AD.
4. Inflammatory Markers and Complement Proteins
It has been found that AD pathology even includes other factors excluding Aβ and NFTs, that are majorly involved in neuronal impairment. The increased expression of pro-inflammatory cytokines in the brain tissues and the blood samples of AD patients confirmed the association of inflammation in the advancement of various diseases including neurodegenerative diseases [118][29]. Angharad et al., 2019, worked on finding a plasma biomarker that aids early diagnosis, stratification, prediction of disease course, or monitoring response to therapy in AD [119][30]. Astrocytes, microglia, cytokines, and chemokines, which are part of the nervous system’s innate immune reaction known as neuroinflammation, are crucial in the pathogenesis of AD’s early stages Figure 1 [120][31]. The accumulated Aβ oligomers stimulate the microglia, initiating the release of pro-inflammatory mediators like glial fibrillary acidic protein (GFAP), neurotoxins, and free radicals [121][32]. These compounds elicit oxidative stress, thereby promoting inflammatory processes in neurons. The MAPK (Mitogen-activated protein kinase) pathway also promotes neurodegeneration by synchronizing with NF-кB (nuclear factor kappa-light-chain-enhancer of activated B cells) increasing pro-inflammatory cytokine production, leading to enhanced APP processing that hyper-phosphorylates the tau protein forming neurofibrillary tangles and eventually collapses the BBB (blood–brain barrier) [122][33]. In AD, exaggerated immune reactivity of p38 MAPK initiates cytokine production via direct phosphorylation of transcription factors and enhanced mRNA translation coding for pro-inflammatory cytokines [123][34]. Once the Aβ peptides accumulate during AD, an immune reaction in the brain initiates the activation and release of inflammatory markers such as TNF-α, IL-1, and IL-6, and the activation of specialized brain cells. Inflammatory markers such as tumor necrosis factor (TNF), interleukin-6 (IL-6), chitinase-3-like protein 1 (CHI3L1 or YKL-40), and acute phase C reactive protein (CRP) were found to have effects on the brains or peripheral regions of dementia patients [124][35]. The activated inflammatory cells further produce complement components within the CNS. In vitro, studies show direct activation of the classical pathways via Aβ1–42 and C1q binding. This C1q binds to Aβ and tau, possibly contributing to neuroinflammation and neurodegeneration [125][36]. Thus, this pro-inflammatory cascade can function as a channel promoting neuroinflammation and neurodegeneration [126][37].
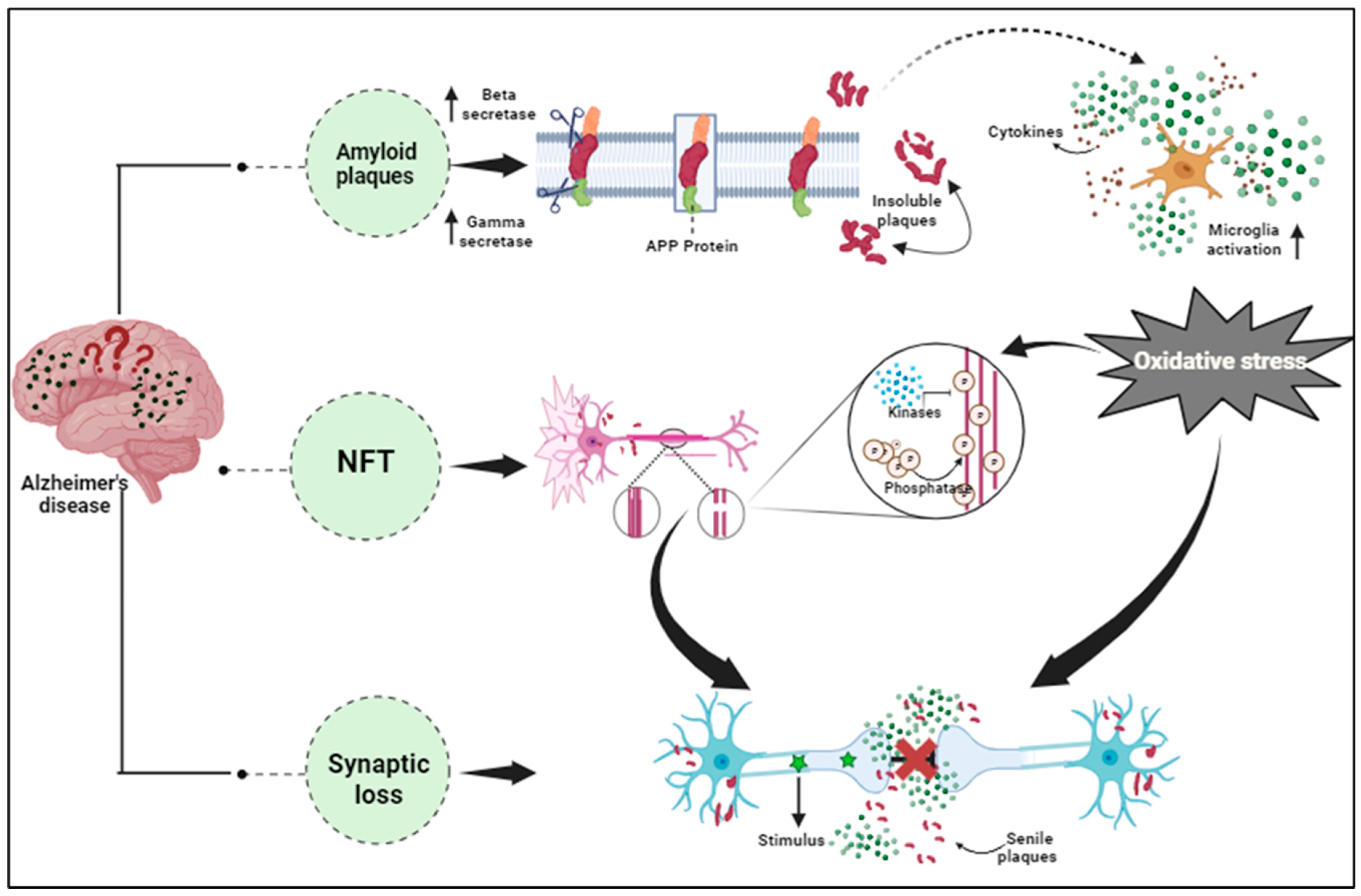
Figure 1.
Pathological hallmarks of Alzheimer’s disease, including amyloid plaques, neurofibrillary tangles, and synaptic dysfunction.
5. Other Potential Proteomic Biomarkers for Alzheimer’s Disease
Apart from the majorly prevalent biomarkers, alpha-Synuclein (α-SN), a pervasive 140 amino acid protein of 18–20 kDa molecular weight is found to be associated with AD. SNCA (Synuclein Alpha), encodes the α-SN, found profusely at the presynaptic terminal of neurons [127][38]. Early-onset AD (EOAD) patients have higher α-SN levels in their CSF, thereby showcasing a probable relation between SNCA gene polymorphisms and AD pathophysiology. α-SN possessing chaperone-like activity might play a role in the regulation of synaptic plasticity, neuronal differentiation, and up-regulation of dopamine release [128][39]. It is primarily associated with synuclein-associated pathologies such as Parkinson’s disease (PD) and dementia with Lewy bodies (DLB), where its misfolding and aggregation play a vital role in the neurodegeneration pathogenesis. DLB is neurodegenerative dementia characterized by the presence of abnormal aggregates of α-SN protein, termed Lewy bodies, in the brain [129][40]. The other associated biomarkers, like the APOEε4 variant, significantly increase the risk of DLB. α-SN deposition or Lewy-related pathology is prominent in AD cases where patients experience visual hallucinations or symptoms that are often associated with Parkinson’s disease. In protein misfolding diseases, the accumulation of α-SN clogs the cellular machinery, disrupting the protein homeostasis network [130][41]. The early stage abnormal α-SN deposition at the presynaptic site in the brain shows its prominent presence in the early events of AD pathogenesis. α-SN directly interacts with Aβ and tau, promoting aggregation, thereby worsening the cognitive decline [131][42]. The abundance of α-SN at the center of Aβ plaques has been verified by immunolabeling using an α-SN antibody, confirming its influence in the formation of Aβ plaques. Increased levels of soluble α-SN monomeric and oligomeric proteins have been found in the temporal region of the AD brain [132][43]. NMR imaging has discovered the selective interaction of the monomeric form of tau with the C-terminal region of the α-SN monomer, thereby elevating its oligomerization and subsequent fibril formation [133][44]. Previous studies have been successful in establishing a relationship between α-SN and AD-associated genes such as APP, PSEN1, and APOE. Elevated α-SN levels in AD could facilitate Aβ oligomerization, tau phosphorylation, activation of kinases, dissociations of tau and tubulin, and tau aggregation. Furthermore, the association of α-SN with genetic factors, such as APP, PSEN1, and APOE, could accelerate AD pathology [134][45]. Apart from this, altered brain protein phosphorylation is a hallmark of AD, and phosphoproteomics offers an opportunity to identify the associated markers. Butterfield et al., 2019, reported two major types of kinases, serine/threonine- and tyrosine-kinases, that catalyzes phosphorylation of serine and threonine and tyrosine residues, respectively, and have a significant role in protein misfolding and aggregation. Along with kinases, protein phosphatases that remove protein-bound phosphate groups were also involved in the phosphorylation of proteins and, consequently, contributed to regulation of protein function [135][46]. Thus, more precise and accurate identification of AD-associated probable factors may help in developing novel therapies or modifying the existing treatments for AD patients.
References
- Dowling, P.; Gargan, S.; Murphy, S.; Zweyer, M.; Sabir, H.; Swandulla, D.; Ohlendieck, K. The Dystrophin Node as Integrator of Cytoskeletal Organization, Lateral Force Transmission, Fiber Stability and Cellular Signaling in Skeletal Muscle. Proteomes 2021, 9, 9.
- Baazaoui, N.; Iqbal, K. Alzheimer’s Disease: Challenges and a Therapeutic Opportunity to Treat It with a Neurotrophic Compound. Biomolecules 2022, 12, 1409.
- Bloom, G.S. Amyloid-β and Tau: The Trigger and Bullet in Alzheimer Disease Pathogenesis. JAMA Neurol. 2014, 71, 505–508.
- Hampel, H.; Hardy, J.; Blennow, K.; Chen, C.; Perry, G.; Kim, S.H.; Villemagne, V.L.; Aisen, P.; Vendruscolo, M.; Iwatsubo, T.; et al. The Amyloid-β Pathway in Alzheimer’s Disease. Mol. Psychiatry 2021, 26, 5481–5503.
- John, A.; Reddy, P.H. Synaptic basis of Alzheimer’s disease: Focus on synaptic amyloid beta, P-tau and mitochondria. Ageing Res. Rev. 2021, 65, 101208.
- Pereira, J.B.; Janelidze, S.; Ossenkoppele, R.; Kvartsberg, H.; Brinkmalm, A.; Mattsson-Carlgren, N.; Stomrud, E.; Smith, R.; Zetterberg, H.; Blennow, K.; et al. Untangling the association of amyloid-β and tau with synaptic and axonal loss in Alzheimer’s disease. Brain 2021, 144, 310–324.
- Li, Y.; Schindler, S.E.; Bollinger, J.G.; Ovod, V.; Mawuenyega, K.G.; Weiner, M.W.; Shaw, L.M.; Masters, C.L.; Fowler, C.J.; Trojanowski, J.Q.; et al. Validation of plasma amyloid-β 42/40 for detecting Alzheimer disease amyloid plaques. Neurology 2022, 98, e688–e699.
- Hansson, O.; Lehmann, S.; Otto, M.; Zetterberg, H.; Lewczuk, P. Advantages and disadvantages of the use of the CSF Amyloid β (Aβ) 42/40 ratio in the diagnosis of Alzheimer’s Disease. Alzheimer’s Res. Ther. 2019, 11, 1–5.
- Lue, L.F.; Guerra, A.; Walker, D.G. Amyloid beta and tau as Alzheimer’s disease blood biomarkers: Promise from new technologies. Neurol. Ther. 2017, 6, 25–36.
- Raulin, A.C.; Doss, S.V.; Trottier, Z.A.; Ikezu, T.C.; Bu, G.; Liu, C.C. ApoE in Alzheimer’s disease: Pathophysiology and therapeutic strategies. Mol. Neurodegener. 2022, 17, 72.
- Kanekiyo, T.; Bu, G. The low-density lipoprotein receptor-related protein 1 and amyloid-β clearance in Alzheimer’s disease. Front. Aging Neurosci. 2014, 6, 93.
- Li, Z.; Shue, F.; Zhao, N.; Shinohara, M.; Bu, G. APOE2: Protective mechanism and therapeutic implications for Alzheimer’s disease. Mol. Neurodegener. 2020, 15, 63.
- Kotze, M.J.; Lückhoff, H.K.; Brand, T.; Pretorius, J.; van Rensburg, S.J. Apolipoprotein E ε-4 as a genetic determinant of Alzheimer’s disease heterogeneity. Degener. Neurol. Neuromuscul. Dis. 2015, 5, 9–18.
- Verghese, P.B.; Castellano, J.M.; Garai, K.; Wang, Y.; Jiang, H.; Shah, A.; Bu, G.; Frieden, C.; Holtzman, D.M. ApoE influences amyloid-β (Aβ) clearance despite minimal apoE/Aβ association in physiological conditions. Proc. Natl. Acad. Sci. USA 2013, 110, E1807–E1816.
- Shi, Y.; Holtzman, D.M. Interplay between innate immunity and Alzheimer disease: APOE and TREM2 in the spotlight. Nat. Rev. Immunol. 2018, 18, 759–772.
- Kurz, C.; Walker, L.; Rauchmann, B.S.; Perneczky, R. Dysfunction of the blood-brain barrier in Alzheimer’s disease: Evidence from human studies. Neuropathol. Appl. Neurobiol. 2022, 48, e12782.
- Milinkeviciute, G.; Green, K.N. Clusterin/apolipoprotein J, its isoforms, and Alzheimer’s disease. Front. Aging Neurosci. 2023, 15, 1167886.
- Liu, Y.; Song, J.H.; Xu, W.; Hou, X.H.; Li, J.Q.; Yu, J.T.; Tan, L.; Chi, S.; Alzheimer’s Disease Neuroimaging Initiative. The associations of cerebrospinal fluid ApoE and biomarkers of Alzheimer’s disease: Exploring interactions with sex. Front. Neurosci. 2021, 15, 633576.
- Lennol, M.P.; Sánchez-Domínguez, I.; Cuchillo-Ibañez, I.; Camporesi, E.; Brinkmalm, G.; Alcolea, D.; Fortea, J.; Lleó, A.; Soria, G.; Aguado, F.; et al. Apolipoprotein E imbalance in the cerebrospinal fluid of Alzheimer’s disease patients. Alzheimer’s Res. Ther. 2022, 14, 1–9.
- Bisht, K.; Sharma, K.; Tremblay, M.È. Chronic stress as a risk factor for Alzheimer’s disease: Roles of microglia-mediated synaptic remodeling, inflammation, and oxidative stress. Neurobio Stress 2018, 9, 9–21.
- Veitch, D.P.; Weiner, M.W.; Aisen, P.S.; Beckett, L.A.; Cairns, N.J.; Green, R.C.; Harvey, D.; Jack, C.R., Jr.; Jagust, W.; Morris, J.C.; et al. Understanding disease progression and improving Alzheimer’s disease clinical trials: Recent highlights from the Alzheimer’s Disease Neuroimaging Initiative. Alzheimer’s Dement. 2019, 15, 106–152.
- Yuste-Checa, P.; Bracher, A.; Hartl, F.U. The chaperone Clusterin in neurodegeneration-friend or foe? Bioessays 2022, 44, e2100287.
- Uddin, M.S.; Kabir, M.T.; Begum, M.M.; Islam, M.S.; Behl, T.; Ashraf, G.M. Exploring the Role of CLU in the Pathogenesis of Alzheimer’s Disease. Neurotox. Res. 2021, 39, 108–2119.
- Li, K.W.; Ganz, A.B.; Smit, A.B. Proteomics of neurodegenerative diseases: Analysis of human post-mortem brain. J. Neurochem. 2019, 151, 435–445.
- Han, S.; Nho, K.; Lee, Y. Alternative Splicing Regulation of an Alzheimer’s Risk Variant in CLU. Int. J. Mol. Sci. 2020, 21, 7079.
- Fareed, M.M.; Qasmi, M.; Aziz, S.; Völker, E.; Förster, C.Y.; Shityakov, S. The Role of Clusterin Transporter in the Pathogenesis of Alzheimer’s Disease at the Blood–Brain Barrier Interface: A Systematic Review. Biomolecules 2022, 12, 1452.
- Foster, E.M.; Dangla-Valls, A.; Lovestone, S.; Ribe, E.M.; Buckley, N.J. Clusterin in Alzheimer’s Disease: Mechanisms, Genetics, and Lessons From Other Pathologies. Front. Neurosci. 2019, 13, 164.
- Yuste-Checa, P.; Trinkaus, V.A.; Riera-Tur, I.; Imamoglu, R.; Schaller, T.F.; Wang, H.; Dudanova, I.; Hipp, M.S.; Bracher, A.; Hartl, F.U. The extracellular chaperone Clusterin enhances Tau aggregate seeding in a cellular model. Nat. Commun. 2021, 12, 4863.
- De Oliveira, J.; Kucharska, E.; Garcez, M.L.; Rodrigues, M.S.; Quevedo, J.; Moreno-Gonzalez, I.; Budni, J. Inflammatory cascade in Alzheimer’s disease pathogenesis: A review of experimental findings. Cells 2021, 28, 2581.
- Morgan, A.R.; Touchard, S.; Leckey, C.; O’Hagan, C.; Nevado-Holgado, A.J.; Barkhof, F.; Bertram, L.; Blin, O.; Bos, I.; Dobricic, V.; et al. Inflammatory biomarkers in Alzheimer’s disease plasma. Alzheimer’s. Dement. 2019, 15, 776–787.
- Webers, A.; Heneka, M.T.; Gleeson, P.A. The role of innate immune responses and neuroinflammation in amyloid accumulation and progression of Alzheimer’s disease. Immunol. Cell Biol. 2020, 98, 28–41.
- Rauf, A.; Badoni, H.; Abu-Izneid, T.; Olatunde, A.; Rahman, M.M.; Painuli, S.; Semwal, P.; Wilairatana, P.; Mubarak, M.S. Neuroinflammatory Markers: Key Indicators in the Pathology of Neurodegenerative Diseases. Molecules 2022, 27, 3194.
- Chavarría, C.; Ivagnes, R.; Souza, J.M. Extracellular Alpha-Synuclein: Mechanisms for Glial Cell Internalization and Activation. Biomolecules 2022, 12, 655.
- Masenga, S.K.; Kabwe, L.S.; Chakulya, M.; Kirabo, A. Mechanisms of Oxidative Stress in Metabolic Syndrome. Int. J. Mol. Sci. 2023, 24, 7898.
- Rather, M.A.; Khan, A.; Alshahrani, S.; Rashid, H.; Qadri, M.; Rashid, S.; Alsaffar, R.M.; Kamal, M.A.; Rehman, M.U. Inflammation and Alzheimer’s Disease: Mechanisms and Therapeutic Implications by Natural Products. Mediat. Inflamm. 2021, 2021, 9982954.
- Shen, X.N.; Niu, L.D.; Wang, Y.J.; Cao, X.P.; Liu, Q.; Tan, L.; Zhang, C.; Yu, J.T. Inflammatory markers in Alzheimer’s disease and mild cognitive impairment: A meta-analysis and systematic review of 170 studies. J. Neurol. Neurosurg. Psychiatry 2019, 90, 590–598.
- Shah, A.; Kishore, U.; Shastri, A. Complement System in Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 13647.
- Agliardi, C.; Guerini, F.R.; Meloni, M.; Clerici, M. Alpha-synuclein as a biomarker in Parkinson’s disease: Focus on neural derived extracelluar vesicles. Neural Reg. Res. 2022, 17, 1503.
- Burré, J.; Sharma, M.; Südhof, T.C. Cell Biology and Pathophysiology of α-Synuclein. Cold Spring Harb. Perspect. Med. 2018, 8, a024091.
- Sengupta, U.; Guerrero-Muñoz, M.J.; Castillo-Carranza, D.L.; Lasagna-Reeves, C.A.; Gerson, J.E.; Paulucci-Holthauzen, A.A.; Krishnamurthy, S.; Farhed, M.; Jackson, G.R.; Kayed, R. Pathological interface between oligomeric alpha-synuclein and tau in synucleinopathies. Biol. Psychiatry 2015, 78, 672–683.
- Sweeney, P.; Park, H.; Baumann, M.; Dunlop, J.; Frydman, J.; Kopito, R.; McCampbell, A.; Leblanc, G.; Venkateswaran, A.; Nurmi, A.; et al. Protein misfolding in neurodegenerative diseases: Implications and strategies. Transl. Neurodegener. 2017, 6, 6.
- Shim, K.H.; Kang, M.J.; Youn, Y.C.; An, S.S.A.; Kim, S. Alpha-synuclein: A pathological factor with Aβ and tau and biomarker in Alzheimer’s disease. Alzheimer’s Res. Ther. 2022, 14, 201.
- Twohig, D.; Nielsen, H.M. α-synuclein in the pathophysiology of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 23.
- Xu, M.M.; Ryan, P.; Rudrawar, S.; Quinn, R.J.; Zhang, H.Y.; Mellick, G.D. Advances in the development of imaging probes and aggregation inhibitors for alpha-synuclein. Acta Pharmacol. Sin. 2020, 41, 483–498.
- Rodriguez-Vieitez, E.; Nielsen, H.M. Associations Between APOE Variants, Tau and α-Synuclein. Adv. Exp. Med. Biol. 2019, 1184, 177–186.
- Butterfield, D.A. Phosphoproteomics of Alzheimer disease brain: Insights into altered brain protein regulation of critical neuronal functions and their contributions to subsequent cognitive loss. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 2031–2039.
More