Organophosphorus compounds (OPs) have applications in agriculture (e.g., pesticides), industry (e.g., flame retardants), and chemical warfare (nerve agents). The main target of OPs is AChE, the enzyme that breaks down ACh into acetic acid and choline, terminating synaptic signal transmission mediated by ACh in neuromuscular junctions, in the autonomic (mainly parasympathetic) nervous system, and in the brain. The OP binds to and phosphorylates a nucleophilic serine at the catalytic site of the enzyme, thus preventing the hydrolysis of ACh and resulting in excessive elevation of ACh in cholinergic synapses. Insects, whose central nervous system utilizes ACh as the major excitatory neurotransmitter, are killed instantly by OPs, primarily due to hyperstimulation of nicotinic cholinergic receptors, which are the most abundant in their central nervous system.
- acetylcholinesterase
- respiratory depression
- neurotoxicity
1. Depression of Respiration
2. Induction of Seizures and Status Epilepticus
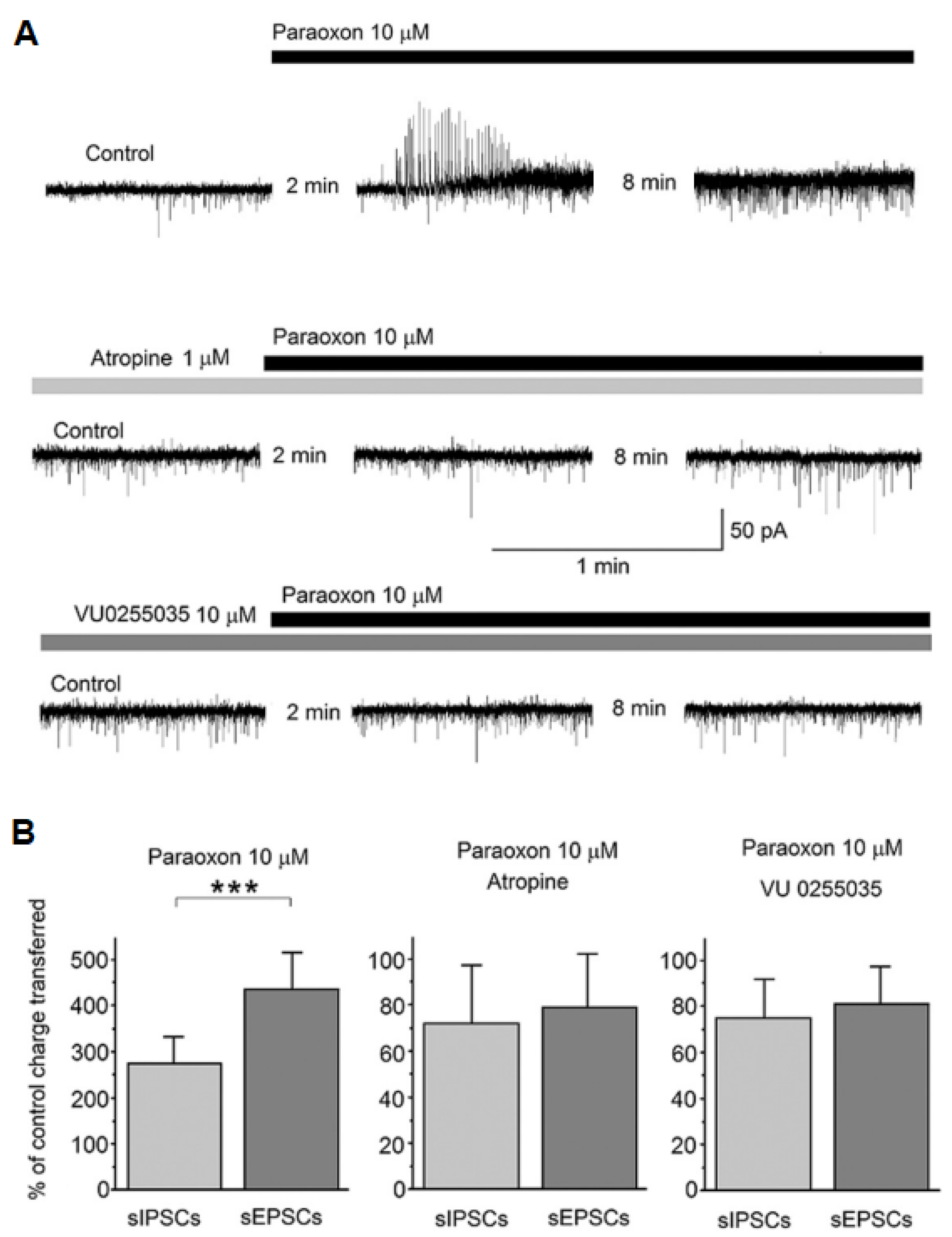
3. Seizures and Status Epilepticus as the Primary Mediators of OP Neurotoxicity
References
- Carey, J.L.; Dunn, C.; Gaspari, R.J. Central respiratory failure during acute organophosphate poisoning. Respir. Physiol. Neurobiol. 2013, 189, 403–410.
- Shao, X.M.; Feldman, J.L. Acetylcholine modulates respiratory pattern: Effects mediated by M3-like receptors in preBötzinger complex inspiratory neurons. J. Neurophysiol. 2000, 83, 1243–1252.
- Lai, J.; Shao, X.M.; Pan, R.W.; Dy, E.; Huang, C.H.; Feldman, J.L. RT-PCR reveals muscarinic acetylcholine receptor mRNA in the pre-Bötzinger complex. Am. J. Physiol. Lung Cell Mol. Physiol. 2001, 281, L1420–L1424.
- Shao, X.M.; Feldman, J.L. Cholinergic neurotransmission in the preBötzinger Complex modulates excitability of inspiratory neurons and regulates respiratory rhythm. Neuroscience 2005, 130, 1069–1081.
- Zheng, F.; Nixdorf-Bergweiler, B.E.; Edelmann, E.; van Brederode, J.F.M.; Alzheimer, C. Muscarinic Modulation of Morphologically Identified Glycinergic Neurons in the Mouse PreBötzinger Complex. Front. Cell Neurosci. 2020, 13, 562.
- Hulse, E.J.; Davies, J.O.; Simpson, A.J.; Sciuto, A.M.; Eddleston, M. Respiratory complications of organophosphorus nerve agent and insecticide poisoning. Implications for respiratory and critical care. Am. J. Respir. Crit. Care Med. 2014, 190, 1342–1354.
- Shao, X.M.; Feldman, J.L. Central cholinergic regulation of respiration: Nicotinic receptors. Acta Pharmacol. Sin. 2009, 30, 761–770.
- Ochoa, E.L.; Chattopadhyay, A.; McNamee, M.G. Desensitization of the nicotinic acetylcholine receptor: Molecular mechanisms and effect of modulators. Cell Mol. Neurobiol. 1989, 9, 141–178.
- Quick, M.W.; Lester, R.A. Desensitization of neuronal nicotinic receptors. J. Neurobiol. 2002, 53, 457–478.
- Paradiso, K.G.; Steinbach, J.H. Nicotine is highly effective at producing desensitization of rat α4β2 neuronal nicotinic receptors. J. Physiol. 2003, 553, 857–871.
- Stewart, W.C.; Anderson, E.A. Effect of a cholinesterase inhibitor when injected into the medulla of the rabbit. J. Pharmacol. Exp. Ther. 1968, 162, 309–318.
- Houze, P.; Pronzola, L.; Kayouka, M.; Villa, A.; Debray, M.; Baud, F.J. Ventilatory effects of low-dose paraoxon result from central muscarinic effects. Toxicol. Appl. Pharmacol. 2008, 233, 186–192.
- Bird, S.B.; Gaspari, R.J.; Dickson, E.W. Early death due to severe organophosphate poisoning is a centrally mediated process. Acad. Emerg. Med. 2003, 10, 295–298.
- Aroniadou-Anderjaska, V.; Apland, J.P.; Figueiredo, T.H.; de Araujo Furtado, M.; Braga, M.F. Acetylcholinesterase inhibitors (nerve agents) as weapons of mass destruction: History, mechanisms of action, and medical countermeasures. Neuropharmacology 2020, 181, 108298.
- Okumura, T.; Takasu, N.; Ishimatsu, S.; Miyanoki, S.; Mitsuhashi, A.; Kumada, K.; Tanaka, K.; Hinohara, S. Report on 640 victims of the Tokyo subway sarin attack. Ann. Emerg. Med. 1996, 28, 129–135.
- Peng, X.; Perkins, M.W.; Simons, J.; Witriol, A.M.; Rodriguez, A.M.; Benjamin, B.M.; Devorak, J.; Sciuto, A.M. Acute pulmonary toxicity following inhalation exposure to aerosolized VX in anesthetized rats. Inhal. Toxicol. 2014, 26, 371–379.
- Figueiredo, T.H.; Apland, J.P.; Braga, M.F.M.; Marini, A.M. Acute and long-term consequences of exposure to organophosphate nerve agents in humans. Epilepsia 2018, 59, 92–99.
- Blum, A.S. Respiratory physiology of seizures. J. Clin. Neurophysiol. 2009, 26, 309–315.
- Dlouhy, B.J.; Gehlbach, B.K.; Kreple, C.J.; Kawasaki, H.; Oya, H.; Buzza, C.; Granner, M.A.; Welsh, M.J.; Howard, M.A.; Wemmie, J.A.; et al. Breathing inhibited when seizures spread to the amygdala and upon amygdala stimulation. J. Neurosci. 2015, 35, 10281–10289.
- Šimić, G.; Tkalčić, M.; Vukić, V.; Mulc, D.; Španić, E.; Šagud, M.; Bordonau, F.E.; Vukšić, M.; R Hof, P. Understanding Emotions: Origins and Roles of the Amygdala. Biomolecules 2021, 11, 823.
- Aroniadou-Anderjaska, V.; Fritsch, B.; Qashu, F.; Braga, M.F. Pathology and pathophysiology of the amygdala in epileptogenesis and epilepsy. Epilepsy Res. 2008, 78, 102–116.
- McDonough, J.H., Jr.; McLeod, C.G., Jr.; Nipwoda, M.T. Direct microinjection of soman or VX into the amygdala produces repetitive limbic convulsions and neuropathology. Brain Res. 1987, 435, 123–137.
- Prager, E.M.; Aroniadou-Anderjaska, V.; Almeida-Suhett, C.P.; Figueiredo, T.H.; Apland, J.P.; Braga, M.F. Acetylcholinesterase inhibition in the basolateral amygdala plays a key role in the induction of status epilepticus after soman exposure. Neurotoxicology 2013, 38, 84–90.
- Lallement, G.; Carpentier, P.; Collet, A.; Pernot-Marino, I.; Baubichon, D.; Sentenac-Roumanou, H.; Blanchet, G. Involvement of glutamatergic system of amygdala in generalized seizures induced by soman: Comparison with the hippocampus. C. R. Acad. Sci. III 1991, 313, 421–426.
- Apland, J.P.; Aroniadou-Anderjaska, V.; Braga, M.F.M. Soman induces ictogenesis in the amygdala and interictal activity in the hippocampus that are blocked by a GluR5 kainate receptor antagonist in vitro. Neuroscience 2009, 159, 380–389.
- Picciotto, M.R.; Higley, M.J.; Mineur, Y.S. Acetylcholine as a neuromodulator: Cholinergic signaling shapes nervous system function and behavior. Neuron 2012, 76, 116–129.
- Kruse, A.C.; Kobilka, B.K.; Gautam, D.; Sexton, P.M.; Christopoulos, A.; Wess, J. Muscarinic acetylcholine receptors: Novel opportunities for drug development. Nat. Rev. Drug. Discov. 2014, 13, 549–560.
- Dani, J.A. Neuronal nicotinic acetylcholine receptor structure and function and response to nicotine. Int. Rev. Neurobiol. 2015, 124, 3–19.
- Thomsen, M.; Sørensen, G.; Dencker, D. Physiological roles of CNS muscarinic receptors gained from knockout mice. Neuropharmacology 2018, 136, 411–420.
- Colangelo, C.; Shichkova, P.; Keller, D.; Markram, H.; Ramaswamy, S. Cellular, Synaptic and Network Effects of Acetylcholine in the Neocortex. Front. Neural Circuits 2019, 13, 24.
- Lallement, G.; Dorandeu, F.; Filliat, P.; Carpentier, P.; Baille, V.; Blanchet, G. Medical management of organophosphate-induced seizures. J. Physiol. Paris 1998, 92, 369–373.
- Shih, T.M.; McDonough, J.H., Jr.; Koplovitz, I. Anticonvulsants for soman-induced seizure activity. J. Biomed. Sci. 1999, 6, 86–96.
- McDonough, J.H., Jr.; Zoeffel, L.D.; McMonagle, J.; Copeland, T.L.; Smith, C.D.; Shih, T.M. Anticonvulsant treatment of nerve agent seizures: Anticholinergics versus diazepam in soman-intoxicated guinea pigs. Epilepsy Res. 2000, 38, 1–14.
- McDonough, J.H., Jr.; Shih, T.M. Neuropharmacological mechanisms of nerve agent-induced seizure and neuropathology. Neurosci. Biobehav. Rev. 1997, 21, 559–579.
- Miller, S.L.; Aroniadou-Anderjaska, V.; Pidoplichko, V.I.; Figueiredo, T.H.; Apland, J.P.; Krishnan, J.K.; Braga, M.F. The M1 muscarinic receptor antagonist VU0255035 delays the development of status epilepticus after organophosphate exposure and prevents hyperexcitability in the basolateral amygdala. J. Pharmacol. Exp. Ther. 2017, 360, 23–32.
- Sheffler, D.J.; Williams, R.; Bridges, T.M.; Xiang, Z.; Kane, A.S.; Byun, N.E.; Jadhav, S.; Mock, M.M.; Zheng, F.; Lewis, L.M.; et al. A novel selective muscarinic acetylcholine receptor subtype 1 antagonist reduces seizures without impairing hippocampus dependent learning. Mol. Pharmacol. 2009, 76, 356–368.
- Fisahn, A.; Yamada, M.; Duttaroy, A.; Gan, J.W.; Deng, C.X.; McBain, C.J.; Wess, J. Muscarinic induction of hippocampal gamma oscillations requires coupling of the M1 receptor to two mixed cation currents. Neuron 2002, 33, 615–624.
- Williamson, J.; Singh, T.; Kapur, J. Neurobiology of organophosphate-induced seizures. Epilepsy Behav. 2019, 101, 106426.
- Aroniadou-Anderjaska, V.; Figueiredo, T.H.; Apland, J.P.; Prager, E.M.; Pidoplichko, V.I.; Miller, S.L.; Braga, M.F. Long-term neuropathological and behavioral impairments after exposure to nerve agents. Ann. N. Y. Acad. Sci. 2016, 1374, 17–28.
- Figueiredo, T.H.; Aroniadou-Anderjaska, V.; Apland, J.P.; Rossetti, K.; Braga, M.F. Delayed tezampanel and caramiphen treatment but not midazolam protects against long-term neuropathology after soman exposure. Exp. Biol. Med. 2023, 248, 612–623.
- de Araujo Furtado, M.; Aroniadou-Anderjaska, V.; Figueiredo, T.H.; Pidoplichko, V.I.; Apland, J.P.; Rossetti, K.; Braga, M.F.M. Preventing Long-Term Brain Damage by Nerve Agent-Induced Status Epilepticus in Rat Models Applicable to Infants: Significant Neuroprotection by Tezampanel Combined with Caramiphen but not by Midazolam Treatment. J. Pharmacol. Exp. Ther. 2023, in press.
- de Araujo Furtado, M.; Lumley, L.A.; Robison, C.; Tong, L.C.; Lichtenstein, S.; Yourick, D.L. Spontaneous recurrent seizures after status epilepticus induced by soman in Sprague-Dawley rats. Epilepsia 2010, 51, 1503–1510.
- Chapman, S.; Yaakov, G.; Egoz, I.; Rabinovitz, I.; Raveh, L.; Kadar, T.; Gilat, E.; Grauer, E. Sarin-induced brain damage in rats is attenuated by delayed administration of midazolam. Neurotoxicology 2015, 49, 132–138.
- Gilat, E.; Kadar, T.; Levy, A.; Rabinovitz, I.; Cohen, G.; Kapon, Y.; Sahar, R.; Brandeis, R. Anticonvulsant treatment of sarin-induced seizures with nasal midazolam: An electrographic, behavioral, and histological study in freely moving rats. Toxicol. Appl. Pharmacol. 2005, 209, 74–85.
- Apland, J.P.; Aroniadou-Anderjaska, V.; Figueiredo, T.H.; de Araujo Furtado, M.; Braga, M.F.M. Full protection against soman-induced seizures and brain damage by LY293558 and caramiphen combination treatment in adult rats. Neurotox. Res. 2018, 34, 511–524.
- Figueiredo, T.H.; Aroniadou-Anderjaska, V.; Pidoplichko, V.I.; Apland, J.P.; Braga, M.F.M. Antiseizure and Neuroprotective Efficacy of Midazolam in Comparison with Tezampanel (LY293558) against Soman-Induced Status Epilepticus. Toxics 2022, 10, 409.
- Shih, T.M.; Duniho, S.M.; McDonough, J.H. Control of nerve agent-induced seizures is critical for neuroprotection and survival. Toxicol. Appl. Pharmacol. 2003, 188, 69–80.
- Tsuchida, T.N.; Barkovich, A.J.; Bollen, A.W.; Hart, A.P.; Ferriero, D.M. Childhood status epilepticus and excitotoxic neuronal injury. Pediatr. Neurol. 2007, 36, 253–257.
- Barker-Haliski, M.; White, H.S. Glutamatergic mechanisms associated with seizures and epilepsy. Cold Spring Harb. Perspect. Med. 2015, 5, 022863.
- Deshpande, L.S.; Carter, D.S.; Blair, R.E.; DeLorenzo, R.J. Development of a prolonged calcium plateau in hippocampal neurons in rats surviving status epilepticus induced by the organophosphate diisopropylfluorophosphate. Toxicol. Sci. 2010, 116, 623–631.
- Fujikawa, D.G. Programmed mechanisms of status epilepticus-induced neuronal necrosis. Epilepsia Open 2023, 8, S25–S34.
- Prathiksha, J.; Narasimhamurthy, R.K.; Dsouza, H.S.; Mumbrekar, K.D. Organophosphate pesticide-induced toxicity through DNA damage and DNA repair mechanisms. Mol. Biol. Rep. 2023, 50, 5465–5479.
- Fink, S.L.; Cookson, B.T. Apoptosis, pyroptosis, and necrosis: Mechanistic description of dead and dying eukaryotic cells. Infect. Immun. 2005, 73, 1907–1916.
- Pinton, P.; Giorgi, C.; Siviero, R.; Zecchini, E.; Rizzuto, R. Calcium and apoptosis: ER-mitochondria Ca2+ transfer in the control of apoptosis. Oncogene 2008, 27, 6407–6418.
- Dingledine, R.; Varvel, N.H.; Dudek, F.E. When and how do seizures kill neurons, and is cell death relevant to epileptogenesis? Adv. Exp. Med. Biol. 2014, 813, 109–122.
- Du, K.; He, M.; Zhao, D.; Wang, Y.; Ma, C.; Liang, H.; Wang, W.; Min, D.; Xue, L.; Guo, F. Mechanism of cell death pathways in status epilepticus and related therapeutic agents. Biomed. Pharmacother. 2022, 149, 112875.
- Niquet, J.; Auvin, S.; Archie, M.; Seo, D.W.; Allen, S.; Sankar, R.; Wasterlain, C.W. Status pilepticus triggers caspase-3 activation and necrosis in the immature rat brain. Epilepsia 2007, 48, 1203–1206.
- Lopez-Meraz, M.L.; Wasterlain, C.G.; Rocha, L.; Allen, S.; Niquet, J. Vulnerability of postnatal hippocampal neurons to seizures varies regionally with their maturational stage. Neurobiol. Dis. 2010, 37, 394–402.
- Niquet, J.; Lopez-Meraz, M.L.; Wasterlain, C.G. Programmed Necrosis After Status Epilepticus. In Jasper’s Basic Mechanisms of the Epilepsies, 4th ed.; Noebels, J.L., Avoli, M., Rogawski, M.A., Olsen, R.W., Delgado-Escueta, A.V., Eds.; National Center for Biotechnology Information (US): Bethesda, MD, USA, 2012; pp. 377–386.
- Ding, S.; Fellin, T.; Zhu, Y.; Lee, S.Y.; Auberson, Y.P.; Meaney, D.F.; Coulter, D.A.; Carmignoto, G.; Haydon, P.G. Enhanced astrocytic Ca2+ signals contribute to neuronal excitotoxicity after status epilepticus. J. Neurosci. 2007, 27, 10674–10684.
- Verkhratsky, A.; Rodríguez, J.J.; Parpura, V. Calcium signalling in astroglia. Mol. Cell Endocrinol. 2012, 353, 45–56.
- Mahmoud, S.; Gharagozloo, M.; Simard, C.; Gris, D. Astrocytes Maintain Glutamate Homeostasis in the CNS by Controlling the Balance between Glutamate Uptake and Release. Cells 2019, 8, 184.
- Vezzani, A.; Aronica, E.; Mazarati, A.; Pittman, Q.J. Epilepsy and brain inflammation. Exp. Neurol. 2013, 244, 11–21.
- Vargas-Sánchez, K.; Mogilevskaya, M.; Rodríguez-Pérez, J.; Rubiano, M.G.; Javela, J.J.; González-Reyes, R.E. Astroglial role in the pathophysiology of status epilepticus: An overview. Oncotarget 2018, 9, 26954–26976.
- Banks, C.N.; Lein, P.J. A review of experimental evidence linking neurotoxic organophosphorus compounds and inflammation. Neurotoxicology 2012, 33, 575–584.
- Pulkrabkova, L.; Svobodova, B.; Konecny, J.; Kobrlova, T.; Muckova, L.; Janousek, J.; Pejchal, J.; Korabecny, J.; Soukup, O. Neurotoxicity evoked by organophosphates and available countermeasures. Arch. Toxicol. 2023, 97, 39–72.
- López-Meraz, M.L.; Álvarez-Croda, D.M. Microglia and status epilepticus in the immature brain. Epilepsia Open 2023, 8, S73–S81.
- Vezzani, A.; French, J.; Bartfai, T.; Baram, T.Z. The role of inflammation in epilepsy. Nat. Rev. Neurol. 2011, 7, 31–40.
- Galic, M.A.; Riazi, K.; Pittman, Q.J. Cytokines and brain excitability. Front. Neuroendocrinol. 2012, 33, 116–125.
- Putra, M.; Sharma, S.; Gage, M.; Gasser, G.; Hinojo-Perez, A.; Olson, A.; Gregory-Flores, A.; Puttachary, S.; Wang, C.; Anantharam, V.; et al. Inducible nitric oxide synthase inhibitor, 1400W, mitigates DFP-induced long-term neurotoxicity in the rat model. Neurobiol. Dis. 2020, 133, 104443.
- Lin, T.K.; Chen, S.D.; Lin, K.J.; Chuang, Y.C. Seizure-Induced Oxidative Stress in Status Epilepticus: Is Antioxidant Beneficial? Antioxidants 2020, 9, 1029.
- Gorter, J.A.; van Vliet, E.A.; Aronica, E. Status epilepticus, blood-brain barrier disruption, inflammation, and epileptogenesis. Epilepsy Behav. 2015, 49, 13–16.
- Mukandala, G.; Tynan, R.; Lanigan, S.; O’Connor, J.J. The Effects of Hypoxia and Inflammation on Synaptic Signaling in the CNS. Brain Sci. 2016, 6, 6.
- Sha, S.; Tan, J.; Miao, Y.; Zhang, Q. The Role of Autophagy in Hypoxia-Induced Neuroinflammation. DNA Cell Biol. 2021, 40, 733–739.
- Guignet, M.; Lein, P.J. Neuroinflammation in organophosphate-induced neurotoxicity. Adv. Neurotoxicol. 2019, 3, 35–79.
- Tian, D.S.; Peng, J.; Murugan, M.; Feng, L.J.; Liu, J.L.; Eyo, U.B.; Zhou, L.J.; Mogilevsky, R.; Wang, W.; Wu, L.J. Chemokine CCL2-CCR2 Signaling Induces Neuronal Cell Death via STAT3 Activation and IL-1β Production after Status Epilepticus. J. Neurosci. 2017, 37, 7878–7892.
- Wolinski, P.; Ksiazek-Winiarek, D.; Glabinski, A. Cytokines and Neurodegeneration in Epileptogenesis. Brain Sci. 2022, 12, 380.
- Rettenbeck, M.L.; von Rüden, E.L.; Bienas, S.; Carlson, R.; Stein, V.M.; Tipold, A.; Potschka, H. Microglial ROS production in an electrical rat post-status epilepticus model of epileptogenesis. Neurosci. Lett. 2015, 599, 146–151.
- Neher, J.J.; Neniskyte, U.; Brown, G.C. Primary phagocytosis of neurons by inflamed microglia: Potential roles in neurodegeneration. Front. Pharmacol. 2012, 3, 27.
- Yanuck, S.F. Microglial Phagocytosis of Neurons: Diminishing Neuronal Loss in Traumatic, Infectious, Inflammatory, and Autoimmune CNS Disorders. Front. Psychiatry 2019, 10, 712.
- Xiong, Z.Q.; Qian, W.; Suzuki, K.; McNamara, J.O. Formation of complement membrane attack complex in mammalian cerebral cortex evokes seizures and neurodegeneration. J. Neurosci. 2003, 23, 955–960.
- Orsini, F.; De Blasio, D.; Zangari, R.; Zanier, E.R.; De Simoni, M.G. Versatility of the complement system in neuroinflammation, neurodegeneration and brain homeostasis. Front. Cell Neurosci. 2014, 8, 380.
- Ziabska, K.; Ziemka-Nalecz, M.; Pawelec, P.; Sypecka, J.; Zalewska, T. Aberrant Complement System Activation in Neurological Disorders. Int. J. Mol. Sci. 2021, 22, 4675.
- Fabene, P.F.; Merigo, F.; Galiè, M.; Benati, D.; Bernardi, P.; Farace, P.; Nicolato, E.; Marzola, P.; Sbarbati, A. Pilocarpine-induced status epilepticus in rats involves ischemic and excitotoxic mechanisms. PLoS ONE 2007, 2, 1105.
- Millis, R.M.; Archer, P.W.; Whittaker, J.A.; Trouth, C.O. The role of hypoxia in organophosphorus nerve agent intoxication. Neurotoxicology 1988, 9, 273–285.