Chronic kidney disease (CKD) is a progressive condition of kidney dysfunction due to diverse causes of injury. In healthy kidneys, protein-bound uremic toxins (PBUTs) are cleared from the systemic circulation by proximal tubule cells through the concerted action of plasma membrane transporters that facilitate their urinary excretion, but the endogenous metabolites are hardly removed with kidney dysfunction and may contribute to CKD progression. Accumulating evidence suggests that senescence of kidney tubule cells influences kidney fibrosis, the common endpoint for CKD with an excessive accumulation of extracellular matrix (ECM). Senescence is a special state of cells characterized by permanent cell cycle arrest and limitation of proliferation, which promotes fibrosis by releasing senescence-associated secretory phenotype (SASP) factors. The accumulation of PBUTs in CKD causes oxidative stress and increases the production of inflammatory (SASP) factors that could trigger fibrosis. Recent sStudies gave some clues that PBUTs may also promote senescence in kidney tubular cells.
- chronic kidney disease
- uremic toxins
- renal tubular transport
- extracellular matrix remodeling
1. Introduction
2. The Mechanisms of Kidney Fibrosis
Kidney fibrosis is induced by the abnormal accumulation of ECM, which often initiates as the result of a wound healing response. The response is orchestrated by complex activities of different cells, including macrophages and T cells, epithelial cells, myofibroblasts, and endothelial cells. Four major phases are involved in this process: (1) primary injury that initiates a fibrotic response; (2) the activation of effector cells, triggering the fibrosis signaling (e.g., TGF-β signaling); (3) production of ECM; and (4) deposition of ECM that promotes tissue fibrosis and eventually leads to kidney failure [1].2.1. Main Signaling of Fibrosis
Three main signaling pathways are involved in fibrosis: transforming growth factor (TGF)-β, wingless/Int (WNT), and yes-associated protein (YAP)/transcriptional coactivator with PDZ-binding motif (TAZ) signaling pathways [14]. TGF-β signals through both canonical (Smad-based) and non-canonical (non-Smad-based) pathways; Smad-based TGF-β signaling plays a central role in the development of renal fibrosis; non-Smad-based profibrotic actions of TGF-β signaling are regulated by interactions with other signaling pathways (e.g., MAPK/ERK and PI3K/AKT pathways signaling) [15]. The WNT signaling pathway is activated by secreted lipid-modified proteins of the WNT family. Activation of WNT signaling stabilizes β-catenin; the nuclear translocation of β-catenin initiates the transcription of fibrotic genes, such as collagen and fibronectin [16,17][16][17]. YAP and TAZ are major players of the Hippo pathway, which is involved in organ development, epithelial homeostasis, tissue regeneration, wound healing, and immune modulation; ECM stiffening promotes the nuclear activity of YAP/TAZ, which in turn promotes the development of a fibrotic cellular phenotype, including increasing the expressions of the connective tissue growth factor (CTGF) and plasminogen activator inhibitor 1 (PAI-1) [18,19,20][18][19][20].2.2. ECM in Kidney Fibrosis
The ECM is a non-cellular component of tissue that provides essential structural support for cellular constituents and acts as an active component in cell signaling. It is composed of water, proteins, and polysaccharides and is responsible for cell–cell communication, cell adhesion, and cell proliferation [23,24][21][22]. There are two main types of ECMs: interstitial connective tissue matrix (e.g., collagen I and fibronectin) and the basement membrane (e.g., collagen IV and laminins) [25][23]. The interstitial connective tissue matrix is responsible for tissue structure, while the basement membrane underlies or surrounds most tissues, including epithelial and endothelial tissues, and interacts with cells (Figure 1) [25,26][23][24].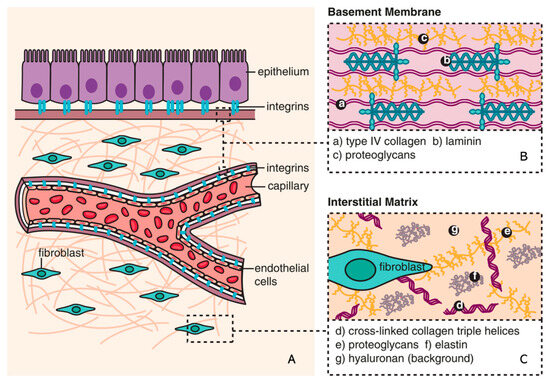
2.3. ECM Remodeling
3. Senescence
Senescence is a special form of permanent cell cycle arrest, which limits cellular proliferation. It was first reported as a loss of replicative capacity in cultured human fibroblasts in 1961 [34][29]. Senescent cells are currently regarded as a potentially important contributor to different types of diseases [35][30], including aging-related diseases [36][31], kidney disease [37][32], and pulmonary disease [38][33]. Some senescent cells can be cleared by immune cells through the chemo-attracting of immune cells, followed by tissue regeneration, which is called acute (short-term) senescence, while chronic (long-term) senescent cells accumulate and create a lesion, aggravating the pathology [39,40][34][35]. Major types of senescence are highlighted as replicative senescence (RS), oncogene-induced senescence (OIS), and stress-induced (premature) senescence (SIS) (Figure 2).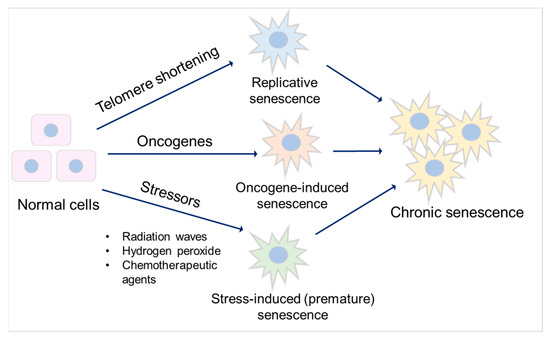
3.1. Mechanisms of Senescence
3.1.1. Cell Cycle Arrest
3.1.2. Apoptosis Resistance
Senescent cells are resistant to apoptosis [61][45] via intrinsic and extrinsic pathways. The intrinsic pathway refers to the mitochondrial pathway of apoptosis, related to mitochondrial outer membrane permeabilization (MOMP) [62][46]. In this pathway, MOMP and the release of cytochrome c are required to trigger apoptosis, and it involves Bcl-2 and caspase family proteins [63,64][47][48]. The Bcl-2 family is divided into three main groups: anti-apoptotic (Bcl-2, Bcl-xl, and Mcl-1), pro-apoptotic (Bax and Bak), and pro-apoptotic BH3-only (Bim, Bid, Bad, and Puma) proteins [65][49]. The balance between pro-apoptotic and anti-apoptotic Bcl-2 family members determines the threshold in MOMP for apoptosis. Caspase proteins are downstream players of MOMP in the intrinsic apoptosis pathway [66][50].3.1.3. SASP Factors
SASP factors are related to a DDR and are generally proinflammatory and/or profibrotic compounds, including numerous cytokines, chemokines, growth factors, and matrix-metalloproteinases (MMPs) [2,72][2][51]. Several reports described that SASP factors are not only responsible for the maintenance and reinforcement of senescence but also key players during its transmission [73][52]. Cytokines, such as IL-6 and IL-8, are well-proven to play such critical roles in stress-induced senescence [74,75,76][53][54][55]. IL-6 maintains senescence through the p53/p21 pathway [77,78][56][57]. This role of IL-6 in senescence is shared by IL-8, which is expressed as a function of IL-6 [75][54]. Both cytokines are regulated by IL-1α [79][58]. The nucleotide-binding oligomerization domain (NOD)-like receptor 3 (NLRP3) inflammasome is upregulated in senescence, which leads to expressions of IL-1α and IL-1β, resulting in the upregulation of SASP factors and the reinforcement of senescence in a paracrine manner [80][59].3.2. Senescence and Fibrosis
Senescence contributes to fibrosis in multiple organs [90,91,92][60][61][62] and is considered to be a result of the release of SASP factors and the pathways triggered by them (Figure 3). TGF-β signaling controls cell proliferation and survival, regulating apoptosis and senescence [87][63], and initiates fibrosis through the canonical Smad signaling and Smad-independent signaling pathways, with subsequent ECM deposition [93][64]. CTGF is the effector molecule of TGF-β in the kidney [94,95][65][66] and has been shown to contribute to TGF-β signaling through the extracellular signal-regulated kinase (ERK), ADAM17, ribosomal S6 kinase 1 (RSK1), and the CCAAT/enhancer-binding protein β (C/EBPβ) signaling pathway in human epithelial cells [85,96][67][68].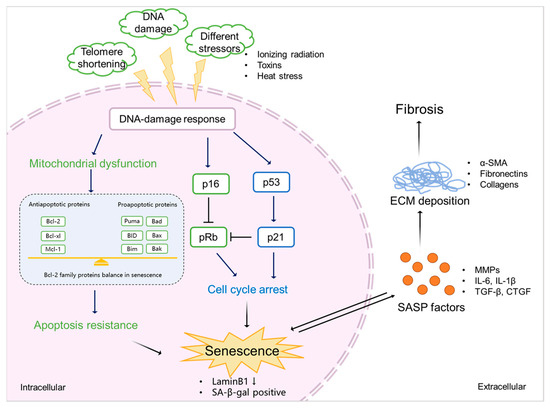
4. Protein-Bound Uremic Toxins Promote Fibrosis by Accelerating Senescence
4.1. PBUTs Accelerate Senescence via Mitochondrial Dysfunction
Different types of senescence have been reported to increase ROS and mitochondrial dysfunction [68][79], which influences the intrinsic apoptosis pathway by the abnormal expression of Bcl-2 family and caspase family markers, which in turn maintain and reinforce senescence [133][80]. Overproduction of ROS during cell stress leads to mitochondrial dysfunction after kidney injury [134][81], which is promoted by PBUT accumulation [135][82]. A cocktail of PBUTs, consisting of IS, PCS, indoxyl-β-glucuronide, p-cresyl glucuronide, indol-3-acetic acid, hippuric acid, kynurenic acid, and l-kynurenine, have been shown to promote ROS production and to upregulate IL-6 in proximal tubule epithelial cells [113,136][78][83].4.2. PBUTs Accelerate Senescence via Cell Cycle Arrest
Cell cycle arrest is necessary for the repair of DNA damage after injury [143][84], which generally occurs in senescence and is a critical factor for fibrosis development [144][85]. DDR is a cause of cell cycle arrest mediated by the p53/p21 and p16/pRb pathways [51][86]. ROS triggers DDR, and DDR promotes ROS production by activating its downstream effectors, including p53 and p21 [145][87].4.3. PBUTs Accelerate Senescence via SASP Factors
During CKD progression, the released inflammatory (SASP) factors activate different pathways and initiate various processes, including senescence and EMT, in tubular epithelial cells [146,149][88][89]. As discussed, PBUT accumulation-induced inflammation might be one reason for senescence development. SASP factors such as IL-6, TGF-β1, and CXCL10 were reported to be increased in proximal tubule cells after the treatment with the PBUTs IS and PCS [142][90].5. Conclusions
PBUTs may promote senescence in CKD through the release of SASP factors (e.g., IL-6 and IL-1β) and common senescence markers (e.g., p21 and Laminb1) and trigger oxidative stress, possibly causing mitochondrial dysfunction, promoting an inflammatory response and increased resistance to cell death. As SASP factors are typically profibrotic and proinflammatory mediators, a novel treatment strategy of CKD could be inhibiting the related signaling, thus suppressing SASP expression. Potential novel agents already exist for this, including anti-fibrotic agents (e.g., TGF-β inhibitor and pirfenidone) and anti-inflammatory agents (e.g., the anti-TNF-α monoclonal antibody and infliximab) [152][91].References
- Rockey, D.C.; Bell, P.D.; Hill, J.A. Fibrosis—A common pathway to organ injury and failure. N. Engl. J. Med. 2015, 372, 1138–1149.
- Hernandez-Segura, A.; Nehme, J.; Demaria, M. Hallmarks of Cellular Senescence. Trends Cell Biol. 2018, 28, 436–453.
- Masereeuw, R. The Dual Roles of Protein-Bound Solutes as Toxins and Signaling Molecules in Uremia. Toxins 2022, 14, 402.
- Maheshwari, V.; Tao, X.; Thijssen, S.; Kotanko, P. Removal of Protein-Bound Uremic Toxins Using Binding Competitors in Hemodialysis: A Narrative Review. Toxins 2021, 13, 622.
- Nigam, S.K.; Bush, K.T. Uraemic syndrome of chronic kidney disease: Altered remote sensing and signalling. Nat. Rev. Nephrol. 2019, 15, 301–316.
- Sun, C.Y.; Chang, S.C.; Wu, M.S. Uremic toxins induce kidney fibrosis by activating intrarenal renin-angiotensin-aldosterone system associated epithelial-to-mesenchymal transition. PLoS ONE 2012, 7, e34026.
- Singh, M.; Yelle, N.; Venugopal, C.; Singh, S.K. EMT: Mechanisms and therapeutic implications. Pharmacol. Ther. 2018, 182, 80–94.
- Faheem, M.M.; Seligson, N.D.; Ahmad, S.M.; Rasool, R.U.; Gandhi, S.G.; Bhagat, M.; Goswami, A. Convergence of therapy-induced senescence (TIS) and EMT in multistep carcinogenesis: Current opinions and emerging perspectives. Cell Death Discov. 2020, 6, 51.
- Kamprom, W.; Tawonsawatruk, T.; Mas-Oodi, S.; Anansilp, K.; Rattanasompattikul, M.; Supokawej, A. P-cresol and Indoxyl Sulfate Impair Osteogenic Differentiation by Triggering Mesenchymal Stem Cell Senescence. Int. J. Med. Sci. 2021, 18, 744–755.
- Kim, S.H.; Yu, M.A.; Ryu, E.S.; Jang, Y.H.; Kang, D.H. Indoxyl sulfate-induced epithelial-to-mesenchymal transition and apoptosis of renal tubular cells as novel mechanisms of progression of renal disease. Lab. Investig. 2012, 92, 488–498.
- Humphreys, B.D.; Lin, S.L.; Kobayashi, A.; Hudson, T.E.; Nowlin, B.T.; Bonventre, J.V.; Valerius, M.T.; McMahon, A.P.; Duffield, J.S. Fate tracing reveals the pericyte and not epithelial origin of myofibroblasts in kidney fibrosis. Am. J. Pathol. 2010, 176, 85–97.
- LeBleu, V.S.; Taduri, G.; O’Connell, J.; Teng, Y.; Cooke, V.G.; Woda, C.; Sugimoto, H.; Kalluri, R. Origin and function of myofibroblasts in kidney fibrosis. Nat. Med. 2013, 19, 1047–1053.
- Zeisberg, E.M.; Potenta, S.E.; Sugimoto, H.; Zeisberg, M.; Kalluri, R. Fibroblasts in kidney fibrosis emerge via endothelial-to-mesenchymal transition. J. Am. Soc. Nephrol. 2008, 19, 2282–2287.
- Piersma, B.; Bank, R.A.; Boersema, M. Signaling in Fibrosis: TGF-beta, WNT, and YAP/TAZ Converge. Front. Med. 2015, 2, 59.
- Meng, X.M.; Nikolic-Paterson, D.J.; Lan, H.Y. TGF-β: The master regulator of fibrosis. Nat. Rev. Nephrol. 2016, 12, 325–338.
- Rim, E.Y.; Clevers, H.; Nusse, R. The Wnt Pathway: From Signaling Mechanisms to Synthetic Modulators. Annu. Rev. Biochem. 2022, 91, 571–598.
- Tan, R.J.; Zhou, D.; Zhou, L.; Liu, Y. Wnt/β-catenin signaling and kidney fibrosis. Kidney Int. Suppl. 2014, 4, 84–90.
- Dey, A.; Varelas, X.; Guan, K.L. Targeting the Hippo pathway in cancer, fibrosis, wound healing and regenerative medicine. Nat. Rev. Drug Discov. 2020, 19, 480–494.
- Liu, F.; Lagares, D.; Choi, K.M.; Stopfer, L.; Marinković, A.; Vrbanac, V.; Probst, C.K.; Hiemer, S.E.; Sisson, T.H.; Horowitz, J.C.; et al. Mechanosignaling through YAP and TAZ drives fibroblast activation and fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 308, L344–L357.
- Zhao, B.; Ye, X.; Yu, J.; Li, L.; Li, W.; Li, S.; Yu, J.; Lin, J.D.; Wang, C.Y.; Chinnaiyan, A.M.; et al. TEAD mediates YAP-dependent gene induction and growth control. Genes. Dev. 2008, 22, 1962–1971.
- Genovese, F.; Manresa, A.A.; Leeming, D.J.; Karsdal, M.A.; Boor, P. The extracellular matrix in the kidney: A source of novel non-invasive biomarkers of kidney fibrosis? Fibrogenesis Tissue Repair 2014, 7, 4.
- Frantz, C.; Stewart, K.M.; Weaver, V.M. The extracellular matrix at a glance. J. Cell Sci. 2010, 123, 4195–4200.
- Bonnans, C.; Chou, J.; Werb, Z. Remodelling the extracellular matrix in development and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 786–801.
- Jayadev, R.; Sherwood, D.R. Basement membranes. Curr. Biol. 2017, 27, R207–R211.
- Ariza de Schellenberger, A.; Bergs, J.; Sack, I.; Taupitz, M. The Extracellular Matrix as a Target for Biophysical and Molecular Magnetic Resonance Imaging. In Quantification of Biophysical Parameters in Medical Imaging; Sack, I., Schaeffter, T., Eds.; Springer International Publishing: Cham, Switzerland, 2018; pp. 123–150.
- Bulow, R.D.; Boor, P. Extracellular Matrix in Kidney Fibrosis: More Than Just a Scaffold. J. Histochem. Cytochem. 2019, 67, 643–661.
- Herrera, J.; Henke, C.A.; Bitterman, P.B. Extracellular matrix as a driver of progressive fibrosis. J. Clin. Investig. 2018, 128, 45–53.
- Peng, W.J.; Yan, J.W.; Wan, Y.N.; Wang, B.X.; Tao, J.H.; Yang, G.J.; Pan, H.F.; Wang, J. Matrix metalloproteinases: A review of their structure and role in systemic sclerosis. J. Clin. Immunol. 2012, 32, 1409–1414.
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621.
- He, S.; Sharpless, N.E. Senescence in Health and Disease. Cell 2017, 169, 1000–1011.
- Childs, B.G.; Durik, M.; Baker, D.J.; van Deursen, J.M. Cellular senescence in aging and age-related disease: From mechanisms to therapy. Nat. Med. 2015, 21, 1424–1435.
- Docherty, M.H.; O’Sullivan, E.D.; Bonventre, J.V.; Ferenbach, D.A. Cellular Senescence in the Kidney. J. Am. Soc. Nephrol. 2019, 30, 726–736.
- Wang, Z.N.; Su, R.N.; Yang, B.Y.; Yang, K.X.; Yang, L.F.; Yan, Y.; Chen, Z.G. Potential Role of Cellular Senescence in Asthma. Front. Cell Dev. Biol. 2020, 8, 59.
- Kobbe, C.v. Targeting senescent cells: Approaches, opportunities, challenges. Aging 2019, 11, 18.
- Munoz-Espin, D.; Serrano, M. Cellular senescence: From physiology to pathology. Nat. Rev. Mol. Cell Biol. 2014, 15, 482–496.
- Liu, X.L.; Ding, J.; Meng, L.H. Oncogene-induced senescence: A double edged sword in cancer. Acta Pharmacol. Sin. 2018, 39, 1553–1558.
- Zhu, H.; Blake, S.; Kusuma, F.K.; Pearson, R.B.; Kang, J.; Chan, K.T. Oncogene-induced senescence: From biology to therapy. Mech. Ageing Dev. 2020, 187, 111229.
- Suzuki, M.; Boothman, D.A. Stress-induced premature senescence (SIPS)—Influence of SIPS on radiotherapy. J. Radiat. Res. 2008, 49, 105–112.
- Kumari, R.; Jat, P. Mechanisms of Cellular Senescence: Cell Cycle Arrest and Senescence Associated Secretory Phenotype. Front. Cell Dev. Biol. 2021, 9, 645593.
- Mijit, M.; Caracciolo, V.; Melillo, A.; Amicarelli, F.; Giordano, A. Role of p53 in the Regulation of Cellular Senescence. Biomolecules 2020, 10, 420.
- Ceccaldi, R.; Parmar, K.; Mouly, E.; Delord, M.; Kim, J.M.; Regairaz, M.; Pla, M.; Vasquez, N.; Zhang, Q.S.; Pondarre, C.; et al. Bone marrow failure in Fanconi anemia is triggered by an exacerbated p53/p21 DNA damage response that impairs hematopoietic stem and progenitor cells. Cell Stem Cell 2012, 11, 36–49.
- Ou, H.L.; Schumacher, B. DNA damage responses and p53 in the aging process. Blood 2018, 131, 488–495.
- Rayess, H.; Wang, M.B.; Srivatsan, E.S. Cellular senescence and tumor suppressor gene p16. Int. J. Cancer 2012, 130, 1715–1725.
- Sperka, T.; Wang, J.; Rudolph, K.L. DNA damage checkpoints in stem cells, ageing and cancer. Nat. Rev. Mol. Cell Biol. 2012, 13, 579–590.
- Childs, B.G.; Baker, D.J.; Kirkland, J.L.; Campisi, J.; van Deursen, J.M. Senescence and apoptosis: Dueling or complementary cell fates? EMBO Rep. 2014, 15, 1139–1153.
- D’Arcy, M.S. Cell death: A review of the major forms of apoptosis, necrosis and autophagy. Cell Biol. Int. 2019, 43, 582–592.
- Ngoi, N.Y.L.; Choong, C.; Lee, J.; Bellot, G.; Wong, A.L.A.; Goh, B.C.; Pervaiz, S. Targeting Mitochondrial Apoptosis to Overcome Treatment Resistance in Cancer. Cancers 2020, 12, 574.
- Van Opdenbosch, N.; Lamkanfi, M. Caspases in Cell Death, Inflammation, and Disease. Immunity 2019, 50, 1352–1364.
- Anantram, A.; Degani, M. Targeting cancer’s Achilles’ heel: Role of BCL-2 inhibitors in cellular senescence and apoptosis. Future Med. Chem. 2019, 11, 2287–2312.
- Shalini, S.; Dorstyn, L.; Dawar, S.; Kumar, S. Old, new and emerging functions of caspases. Cell Death Differ. 2015, 22, 526–539.
- Birch, J.; Gil, J. Senescence and the SASP: Many therapeutic avenues. Genes. Dev. 2020, 34, 1565–1576.
- Kuilman, T.; Peeper, D.S. Senescence-messaging secretome: SMS-ing cellular stress. Nat. Rev. Cancer 2009, 9, 81–94.
- Acosta, J.C.; O’Loghlen, A.; Banito, A.; Guijarro, M.V.; Augert, A.; Raguz, S.; Fumagalli, M.; Da Costa, M.; Brown, C.; Popov, N.; et al. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell 2008, 133, 1006–1018.
- Kuilman, T.; Michaloglou, C.; Vredeveld, L.C.; Douma, S.; van Doorn, R.; Desmet, C.J.; Aarden, L.A.; Mooi, W.J.; Peeper, D.S. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell 2008, 133, 1019–1031.
- You, K.; Parikh, P.; Khandalavala, K.; Wicher, S.A.; Manlove, L.; Yang, B.; Roesler, A.; Roos, B.B.; Teske, J.J.; Britt, R.D., Jr.; et al. Moderate hyperoxia induces senescence in developing human lung fibroblasts. Am. J. Physiol. Lung Cell. Mol. Physiol. 2019, 317, L525–L536.
- Li, Y.; Lu, L.; Xie, Y.; Chen, X.; Tian, L.; Liang, Y.; Li, H.; Zhang, J.; Liu, Y.; Yu, X. Interleukin-6 Knockout Inhibits Senescence of Bone Mesenchymal Stem Cells in High-Fat Diet-Induced Bone Loss. Front. Endocrinol. 2020, 11, 622950.
- Effenberger, T.; von der Heyde, J.; Bartsch, K.; Garbers, C.; Schulze-Osthoff, K.; Chalaris, A.; Murphy, G.; Rose-John, S.; Rabe, B. Senescence-associated release of transmembrane proteins involves proteolytic processing by ADAM17 and microvesicle shedding. FASEB J. 2014, 28, 4847–4856.
- Orjalo, A.V.; Bhaumik, D.; Gengler, B.K.; Scott, G.K.; Campisi, J. Cell surface-bound IL-1alpha is an upstream regulator of the senescence-associated IL-6/IL-8 cytokine network. Proc. Natl. Acad. Sci. USA 2009, 106, 17031–17036.
- Acosta, J.C.; Banito, A.; Wuestefeld, T.; Georgilis, A.; Janich, P.; Morton, J.P.; Athineos, D.; Kang, T.W.; Lasitschka, F.; Andrulis, M.; et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat. Cell Biol. 2013, 15, 978–990.
- Hernandez-Gonzalez, F.; Faner, R.; Rojas, M.; Agustí, A.; Serrano, M.; Sellarés, J. Cellular Senescence in Lung Fibrosis. Int. J. Mol. Sci. 2021, 22, 7012.
- Zhang, M.; Serna-Salas, S.; Damba, T.; Borghesan, M.; Demaria, M.; Moshage, H. Hepatic stellate cell senescence in liver fibrosis: Characteristics, mechanisms and perspectives. Mech. Ageing Dev. 2021, 199, 111572.
- Chen, M.S.; Lee, R.T.; Garbern, J.C. Senescence mechanisms and targets in the heart. Cardiovasc. Res. 2022, 118, 1173–1187.
- Zhang, Y.; Alexander, P.B.; Wang, X.F. TGF-β Family Signaling in the Control of Cell Proliferation and Survival. Cold Spring Harb. Perspect. Biol. 2017, 9, a022145.
- Kim, K.K.; Sheppard, D.; Chapman, H.A. TGF-β1 Signaling and Tissue Fibrosis. Cold Spring Harb. Perspect. Biol. 2018, 10, a022293.
- Samarakoon, R.; Dobberfuhl, A.D.; Cooley, C.; Overstreet, J.M.; Patel, S.; Goldschmeding, R.; Meldrum, K.K.; Higgins, P.J. Induction of renal fibrotic genes by TGF-β1 requires EGFR activation, p53 and reactive oxygen species. Cell Signal. 2013, 25, 2198–2209.
- Ramazani, Y.; Knops, N.; Elmonem, M.A.; Nguyen, T.Q.; Arcolino, F.O.; van den Heuvel, L.; Levtchenko, E.; Kuypers, D.; Goldschmeding, R. Connective tissue growth factor (CTGF) from basics to clinics. Matrix Biol. 2018, 68–69, 44–66.
- Ou, S.C.; Bai, K.J.; Cheng, W.H.; Chen, J.Y.; Lin, C.H.; Wen, H.C.; Chen, B.C. TGF-β Induced CTGF Expression in Human Lung Epithelial Cells through ERK, ADAM17, RSK1, and C/EBPβ Pathways. Int. J. Mol. Sci. 2020, 21, 9084.
- Yanagihara, T.; Tsubouchi, K.; Gholiof, M.; Chong, S.G.; Lipson, K.E.; Zhou, Q.; Scallan, C.; Upagupta, C.; Tikkanen, J.; Keshavjee, S.; et al. Connective-Tissue Growth Factor Contributes to TGF-β1-induced Lung Fibrosis. Am. J. Respir. Cell Mol. Biol. 2022, 66, 260–270.
- Zhang, S.; Fan, Y.; Qin, L.; Fang, X.; Zhang, C.; Yue, J.; Bai, W.; Wang, G.; Chen, Z.; Renz, H.; et al. IL-1β augments TGF-β inducing epithelial-mesenchymal transition of epithelial cells and associates with poor pulmonary function improvement in neutrophilic asthmatics. Respir. Res. 2021, 22, 216.
- Wilson, M.S.; Madala, S.K.; Ramalingam, T.R.; Gochuico, B.R.; Rosas, I.O.; Cheever, A.W.; Wynn, T.A. Bleomycin and IL-1beta-mediated pulmonary fibrosis is IL-17A dependent. J. Exp. Med. 2010, 207, 535–552.
- Fielding, C.A.; Jones, G.W.; McLoughlin, R.M.; McLeod, L.; Hammond, V.J.; Uceda, J.; Williams, A.S.; Lambie, M.; Foster, T.L.; Liao, C.T.; et al. Interleukin-6 signaling drives fibrosis in unresolved inflammation. Immunity 2014, 40, 40–50.
- Epstein Shochet, G.; Brook, E.; Bardenstein-Wald, B.; Shitrit, D. TGF-β pathway activation by idiopathic pulmonary fibrosis (IPF) fibroblast derived soluble factors is mediated by IL-6 trans-signaling. Respir. Res. 2020, 21, 56.
- Levi, N.; Papismadov, N.; Solomonov, I.; Sagi, I.; Krizhanovsky, V. The ECM path of senescence in aging: Components and modifiers. FEBS J. 2020, 287, 2636–2646.
- Vanholder, R.; De Smet, R.; Glorieux, G.; Argilés, A.; Baurmeister, U.; Brunet, P.; Clark, W.; Cohen, G.; De Deyn, P.P.; Deppisch, R.; et al. Review on uremic toxins: Classification, concentration, and interindividual variability. Kidney Int. 2003, 63, 1934–1943.
- Rocchetti, M.T.; Cosola, C.; Ranieri, E.; Gesualdo, L. Protein-bound uremic toxins and immunity. In Cytotoxic T-Cells: Methods and Protocols; Gigante, M., Ranieri, E., Eds.; Springer: New York, NY, USA, 2021; pp. 215–227.
- Fujii, H.; Goto, S.; Fukagawa, M. Role of Uremic Toxins for Kidney, Cardiovascular, and Bone Dysfunction. Toxins 2018, 10, 202.
- Chao, C.T.; Chiang, C.K. Uremic toxins, oxidative stress, and renal fibrosis: An interwined complex. J. Ren. Nutr. 2015, 25, 155–159.
- Mihajlovic, M.; Krebber, M.M.; Yang, Y.; Ahmed, S.; Lozovanu, V.; Andreeva, D.; Verhaar, M.C.; Masereeuw, R. Protein-Bound Uremic Toxins Induce Reactive Oxygen Species-Dependent and Inflammasome-Mediated IL-1beta Production in Kidney Proximal Tubule Cells. Biomedicines 2021, 9, 1326.
- Korolchuk, V.I.; Miwa, S.; Carroll, B.; von Zglinicki, T. Mitochondria in Cell Senescence: Is Mitophagy the Weakest Link? eBioMedicine 2017, 21, 7–13.
- Miwa, S.; Kashyap, S.; Chini, E.; von Zglinicki, T. Mitochondrial dysfunction in cell senescence and aging. J. Clin. Investig. 2022, 132, e158447.
- Zhao, M.; Wang, Y.; Li, L.; Liu, S.; Wang, C.; Yuan, Y.; Yang, G.; Chen, Y.; Cheng, J.; Lu, Y.; et al. Mitochondrial ROS promote mitochondrial dysfunction and inflammation in ischemic acute kidney injury by disrupting TFAM-mediated mtDNA maintenance. Theranostics 2021, 11, 1845–1863.
- Rossi, M.; Campbell, K.L.; Johnson, D.W.; Stanton, T.; Vesey, D.A.; Coombes, J.S.; Weston, K.S.; Hawley, C.M.; McWhinney, B.C.; Ungerer, J.P.; et al. Protein-bound uremic toxins, inflammation and oxidative stress: A cross-sectional study in stage 3-4 chronic kidney disease. Arch. Med. Res. 2014, 45, 309–317.
- Mihajlovic, M.; Fedecostante, M.; Oost, M.J.; Steenhuis, S.K.P.; Lentjes, E.; Maitimu-Smeele, I.; Janssen, M.J.; Hilbrands, L.B.; Masereeuw, R. Role of Vitamin D in Maintaining Renal Epithelial Barrier Function in Uremic Conditions. Int. J. Mol. Sci. 2017, 18, 2531.
- Branzei, D.; Foiani, M. Regulation of DNA repair throughout the cell cycle. Nat. Rev. Mol. Cell Biol. 2008, 9, 297–308.
- Yang, L.; Besschetnova, T.Y.; Brooks, C.R.; Shah, J.V.; Bonventre, J.V. Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat. Med. 2010, 16, 535–543.
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078.
- Passos, J.F.; Nelson, G.; Wang, C.; Richter, T.; Simillion, C.; Proctor, C.J.; Miwa, S.; Olijslagers, S.; Hallinan, J.; Wipat, A.; et al. Feedback between p21 and reactive oxygen production is necessary for cell senescence. Mol. Syst. Biol. 2010, 6, 347.
- Yang, Y.; Mihajlovic, M.; Janssen, M.J.; Masereeuw, R. The Uremic Toxin Indoxyl Sulfate Accelerates Senescence in Kidney Proximal Tubule Cells. Toxins 2023, 15, 242.
- Humphreys, B.D. Mechanisms of Renal Fibrosis. Annu. Rev. Physiol. 2018, 80, 309–326.
- Sun, C.Y.; Hsu, H.H.; Wu, M.S. p-Cresol sulfate and indoxyl sulfate induce similar cellular inflammatory gene expressions in cultured proximal renal tubular cells. Nephrol. Dial. Transplant. 2013, 28, 70–78.
- Yan, M.T.; Chao, C.T.; Lin, S.H. Chronic Kidney Disease: Strategies to Retard Progression. Int. J. Mol. Sci. 2021, 22, 10084.