Kidney fibrosis leads to organ failure by an excessive accumulation of extracellular matrix (ECM), which is the common endpoint for a variety of progressive chronic kidney diseases (CKD)
[1]. Senescence is a special form of permanent cell cycle arrest, which limits proliferation and is highly related to inflammation and fibrosis. Senescent cells exacerbate these processes by releasing senescence-associated secretory phenotype (SASP) factors, which are of pro-inflammatory and profibrotic nature
[2]. Uremic toxins are metabolites that accumulate during kidney disease. Protein-bound uremic toxins (PBUTs) are mostly less than 500 Da but are poorly removed with kidney dysfunction, as they are tightly bound to plasma proteins and can also hardly cross dialyzer membranes
[3][4][3,4]. PBUTs, such as indoxyl sulfate (IS) and p-cresol sulfate (PCS), accumulate in CKD, maintaining and reinforcing CKD and kidney fibrosis
[5][6][5,6]. Recent studies reported that IS and PCS activate the renal RAAS/TGF-β pathway and induce epithelial mesenchymal transition (EMT)
[6]. EMT is a common process during fibrosis and concerns the loss of a differentiated epithelial-like state of cells (e.g., cell-to-cell junctions) to acquire a more mesenchymal-like phenotype (e.g., enhanced ECM expression)
[7]. Senescence and EMT are both characterized by cell dedifferentiation, loss of epithelial phenotype, cell cycle arrest, and negative effects on surrounding cells
[8]. IS triggers senescence
[9] and induces EMT with ECM (i.e., α-SMA) deposition in vitro
[10], which suggests that PBUTs may induce kidney fibrosis by propagating senescence. However, the crosstalk between PBUT-related fibrosis and senescence-related fibrosis remains unclear. It is worth noting that EMT can be induced by toxins, such as IS, in vitro in epithelial cells, but such an EMT mechanism in vivo in renal fibrosis is still questionable, as pericytes, endothelial cells, and bone marrow-derived stem cells may be sources of myofibroblasts
[11][12][13][11,12,13].
2. The Mechanisms of Kidney Fibrosis
Kidney fibrosis is induced by the abnormal accumulation of ECM, which often initiates as the result of a wound healing response. The response is orchestrated by complex activities of different cells, including macrophages and T cells, epithelial cells, myofibroblasts, and endothelial cells. Four major phases are involved in this process: (1) primary injury that initiates a fibrotic response; (2) the activation of effector cells, triggering the fibrosis signaling (e.g., TGF-β signaling); (3) production of ECM; and (4) deposition of ECM that promotes tissue fibrosis and eventually leads to kidney failure
[1].
2.1. Main Signaling of Fibrosis
Three main signaling pathways are involved in fibrosis: transforming growth factor (TGF)-β, wingless/Int (WNT), and yes-associated protein (YAP)/transcriptional coactivator with PDZ-binding motif (TAZ) signaling pathways
[14]. TGF-β signals through both canonical (Smad-based) and non-canonical (non-Smad-based) pathways; Smad-based TGF-β signaling plays a central role in the development of renal fibrosis; non-Smad-based profibrotic actions of TGF-β signaling are regulated by interactions with other signaling pathways (e.g., MAPK/ERK and PI3K/AKT pathways signaling)
[15]. The WNT signaling pathway is activated by secreted lipid-modified proteins of the WNT family. Activation of WNT signaling stabilizes β-catenin; the nuclear translocation of β-catenin initiates the transcription of fibrotic genes, such as collagen and fibronectin
[16][17][16,17]. YAP and TAZ are major players of the Hippo pathway, which is involved in organ development, epithelial homeostasis, tissue regeneration, wound healing, and immune modulation; ECM stiffening promotes the nuclear activity of YAP/TAZ, which in turn promotes the development of a fibrotic cellular phenotype, including increasing the expressions of the connective tissue growth factor (CTGF) and plasminogen activator inhibitor 1 (PAI-1)
[18][19][20][18,19,20].
2.2. ECM in Kidney Fibrosis
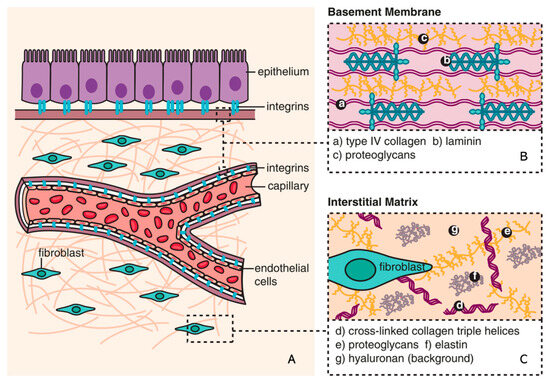
The ECM is a non-cellular component of tissue that provides essential structural support for cellular constituents and acts as an active component in cell signaling. It is composed of water, proteins, and polysaccharides and is responsible for cell–cell communication, cell adhesion, and cell proliferation
[21][22][23,24]. There are two main types of ECMs: interstitial connective tissue matrix (e.g., collagen I and fibronectin) and the basement membrane (e.g., collagen IV and laminins)
[23][25]. The interstitial connective tissue matrix is responsible for tissue structure, while the basement membrane underlies or surrounds most tissues, including epithelial and endothelial tissues, and interacts with cells (
Figure 1)
[23][24][25,26].
Figure 1. Composition of ECM (reproduced with permission from
[25][28]). (
A) The basic subdivision of the ECM into the (
B) basement membrane and (
C) interstitial matrix is shown along with major structural components (collagen and elastin), as well as the background matrix made up of proteoglycans and hyaluronan
[25][28].
2.3. ECM Remodeling
ECM remodeling is referred to as a balance between degradation and production of ECM. When the balance is disrupted
[26][27], a positive feedback loop resulting in increased ECM production drives the development of fibrosis
[27][29]. The cleavage of ECM by different proteases is the main process during the remodeling and includes matrix metalloproteinases (MMPs), adamalysins, meprins, and metalloproteinase inhibitors (reviewed in
[23][25]). MMPs are the main enzymes involved in ECM degradation and remodeling. MMPs can cleave ECM components and activate other MMPs and proteins. Various cytokines (interleukin [IL] and tumor necrosis factor [TNF]) and growth factors (epidermal growth factor [EGF] and transforming growth factor [TGF]) may be involved in the gene expression of MMPs at the transcription level
[28][30].
3. Senescence
Senescence is a special form of permanent cell cycle arrest, which limits cellular proliferation. It was first reported as a loss of replicative capacity in cultured human fibroblasts in 1961
[29][34]. Senescent cells are currently regarded as a potentially important contributor to different types of diseases
[30][35], including aging-related diseases
[31][36], kidney disease
[32][37], and pulmonary disease
[33][38]. Some senescent cells can be cleared by immune cells through the chemo-attracting of immune cells, followed by tissue regeneration, which is called acute (short-term) senescence, while chronic (long-term) senescent cells accumulate and create a lesion, aggravating the pathology
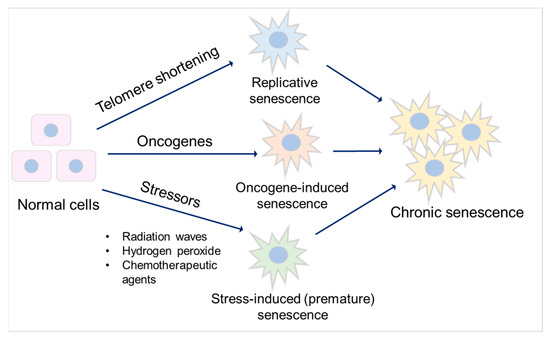
[34][35][39,40]. Major types of senescence are highlighted as replicative senescence (RS), oncogene-induced senescence (OIS), and stress-induced (premature) senescence (SIS) (
Figure 2).
Figure 2. Major types of senescence. Three main types of senescence are identified. Replicative senescence links to telomere shortening that is associated with cell division. Oncogene-induced senescence refers to cell cycle arrest by the aberrant activation of oncogenic signaling, which promotes the initiation and development of cancer. Stress-induced (premature) senescence appears after exposing cells to chemical or physical stresses. Accumulation of long-term senescent cells leads to chronic senescence.
Oncogene-induced senescence refers to cell cycle arrest by the aberrant activation of oncogenic signaling, which promotes the initiation and development of cancer
[36][45]. This can be caused by numerous oncogenes, including constitutively active variants in the RAS/MAPK pathway (RAS-induced senescence), as well as in the PI3K/AKT pathway (AKT-induced senescence). The former undergoes a DDR, while the latter is independent of DDR
[37][46]. Stress-induced (premature) senescence appears after exposing cells to chemical or physical stresses, including radiation waves, hydrogen peroxide, and chemotherapeutic agents
[38][47], leading to cellular stress, increased reactive oxygen species (ROS) generation, and subsequent DNA damage, eventually contributing to senescence
[35][38][40,47].
3.1. Mechanisms of Senescence
3.1.1. Cell Cycle Arrest
Cell cycle arrest in senescence is largely mediated via the p53/p21
CIP1/WAF1 (p21) and p16
Ink4a (p16)/pRb checkpoint pathways controlled by DDR
[39][40][52,53], which are independent processes in senescence induction. p53/p21 is activated when DDR occurs, promoting a p21-dependent G0/G1 cell cycle arrest
[41][42][54,55]. p16 suppresses retinoblastoma 1 (pRb) and prevents the actions of the cyclin-dependent kinases, which induces a G1 cell cycle arrest
[43][56]. Acute DNA damage drives cell cycle arrest via the p53/p21 pathway, while chronic DNA damage followed by the induction of the p16/pRB pathway maintains cell cycle arrest and senescence
[44][57].
3.1.2. Apoptosis Resistance
Senescent cells are resistant to apoptosis
[45][61] via intrinsic and extrinsic pathways. The intrinsic pathway refers to the mitochondrial pathway of apoptosis, related to mitochondrial outer membrane permeabilization (MOMP)
[46][62]. In this pathway, MOMP and the release of cytochrome c are required to trigger apoptosis, and it involves Bcl-2 and caspase family proteins
[47][48][63,64]. The Bcl-2 family is divided into three main groups: anti-apoptotic (Bcl-2, Bcl-xl, and Mcl-1), pro-apoptotic (Bax and Bak), and pro-apoptotic BH3-only (Bim, Bid, Bad, and Puma) proteins
[49][65]. The balance between pro-apoptotic and anti-apoptotic Bcl-2 family members determines the threshold in MOMP for apoptosis. Caspase proteins are downstream players of MOMP in the intrinsic apoptosis pathway
[50][66].
3.1.3. SASP Factors
SASP factors are related to a DDR and are generally proinflammatory and/or profibrotic compounds, including numerous cytokines, chemokines, growth factors, and matrix-metalloproteinases (MMPs)
[2][51][2,72]. Several reports described that SASP factors are not only responsible for the maintenance and reinforcement of senescence but also key players during its transmission
[52][73]. Cytokines, such as IL-6 and IL-8, are well-proven to play such critical roles in stress-induced senescence
[53][54][55][74,75,76]. IL-6 maintains senescence through the p53/p21 pathway
[56][57][77,78]. This role of IL-6 in senescence is shared by IL-8, which is expressed as a function of IL-6
[54][75]. Both cytokines are regulated by IL-1α
[58][79]. The nucleotide-binding oligomerization domain (NOD)-like receptor 3 (NLRP3) inflammasome is upregulated in senescence, which leads to expressions of IL-1α and IL-1β, resulting in the upregulation of SASP factors and the reinforcement of senescence in a paracrine manner
[59][80].
3.2. Senescence and Fibrosis
Senescence contributes to fibrosis in multiple organs
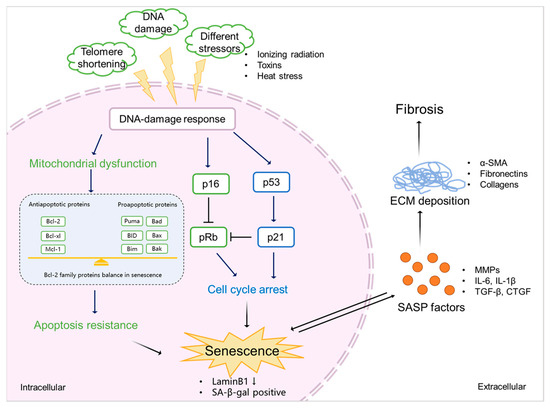
[60][61][62][90,91,92] and is considered to be a result of the release of SASP factors and the pathways triggered by them (
Figure 3). TGF-β signaling controls cell proliferation and survival, regulating apoptosis and senescence
[63][87], and initiates fibrosis through the canonical Smad signaling and Smad-independent signaling pathways, with subsequent ECM deposition
[64][93]. CTGF is the effector molecule of TGF-β in the kidney
[65][66][94,95] and has been shown to contribute to TGF-β signaling through the extracellular signal-regulated kinase (ERK), ADAM17, ribosomal S6 kinase 1 (RSK1), and the CCAAT/enhancer-binding protein β (C/EBPβ) signaling pathway in human epithelial cells
[67][68][85,96].
Figure 3. The mechanism of senescence in kidney fibrosis. Senescence is initiated by various stimulations (e.g., ionizing radiation, exposure to toxins, and heat stress), which triggers DDR. This, on the one hand, induces mitochondrial dysfunction, resulting in the abnormal expression of Bcl-2 family proteins, eventually leading to apoptosis resistance and the promotion of senescence. On the other hand, DDR mediates cell cycle arrest via p53/p21 and p16/pRb checkpoint pathways, which also results in senescence. Senescent cells show a downregulation of LaminB1 and SA-β-gal. SASP factors, including profibrotic cytokines (TGF-β and CTGF), proinflammatory cytokines (IL-6 and IL-1β), and ECM-remodeling proteases (MMPs) expressed by senescent cells promote ECM deposition (α-SMA, fibronectins, and collagens), finally leading to kidney fibrosis.
Proinflammatory mediators such as IL-1β and IL-6 are also involved in fibrosis. IL-1β augments TGF-β1-induced EMT through MAPK signaling pathways
[69][98], which may be dependent on IL-17A
[70][99]. IL-6 shifts acute inflammation into a chronic fibrosis state by regulating MMPs and the TGF-β pathway
[71][72][100,101]. MMPs release ectodomains of cell surface receptors and activate other SASP factors
[73][89], thus regulating ECM production and promoting EMT and kidney fibrosis
[23][25].
4. Protein-Bound Uremic Toxins Promote Fibrosis by Accelerating Senescence
Uremic toxins are endogenous metabolites that are excreted into the urine through glomerular filtration and active transport by the proximal epithelial cells
[74][105]. In kidney disease, uremic toxins management is compromised, which leads to the systemic accumulation of the toxins and activation of inflammation and oxidative stress. Furthermore, uremic toxins can induce profibrotic effects, promoting the progression of kidney damage
[75][106]. Uremic toxins are divided into three distinct groups: (1) small water-soluble compounds (molecular weight <500 Da, e.g., creatinine, urea, and uric acid); (2) middle molecules (peptides with molecular weight >500 Da, e.g., IL-6, IL-8 and TNF-α); and (3) protein-bound uremic toxins (PBUTs; molecular weight mostly <500 Da, e.g., indoxyl sulfate, p-cresyl sulfate, and p-cresol)
[74][76][105,107].
PBUTs accumulate systemically but also in kidney tissue, where they can induce oxidative stress and stimulate the production of inflammatory factors, which might be a trigger for fibrosis
[77][112]. PBUTs induce ROS production and enhance oxidative stress and IL-1β (SASP) expression in kidney proximal tubule cells
[78][113].
4.1. PBUTs Accelerate Senescence via Mitochondrial Dysfunction
Different types of senescence have been reported to increase ROS and mitochondrial dysfunction
[79][68], which influences the intrinsic apoptosis pathway by the abnormal expression of Bcl-2 family and caspase family markers, which in turn maintain and reinforce senescence
[80][133]. Overproduction of ROS during cell stress leads to mitochondrial dysfunction after kidney injury
[81][134], which is promoted by PBUT accumulation
[82][135]. A cocktail of PBUTs, consisting of IS, PCS, indoxyl-β-glucuronide, p-cresyl glucuronide, indol-3-acetic acid, hippuric acid, kynurenic acid, and l-kynurenine, have been shown to promote ROS production and to upregulate IL-6 in proximal tubule epithelial cells
[78][83][113,136].
4.2. PBUTs Accelerate Senescence via Cell Cycle Arrest
Cell cycle arrest is necessary for the repair of DNA damage after injury
[84][143], which generally occurs in senescence and is a critical factor for fibrosis development
[85][144]. DDR is a cause of cell cycle arrest mediated by the p53/p21 and p16/pRb pathways
[86][51]. ROS triggers DDR, and DDR promotes ROS production by activating its downstream effectors, including p53 and p21
[87][145].
4.3. PBUTs Accelerate Senescence via SASP Factors
During CKD progression, the released inflammatory (SASP) factors activate different pathways and initiate various processes, including senescence and EMT, in tubular epithelial cells
[88][89][146,149]. As discussed, PBUT accumulation-induced inflammation might be one reason for senescence development. SASP factors such as IL-6, TGF-β1, and CXCL10 were reported to be increased in proximal tubule cells after the treatment with the PBUTs IS and PCS
[90][142].
5. Conclusions
PBUTs may promote senescence in CKD through the release of SASP factors (e.g., IL-6 and IL-1β) and common senescence markers (e.g., p21 and Laminb1) and trigger oxidative stress, possibly causing mitochondrial dysfunction, promoting an inflammatory response and increased resistance to cell death. As SASP factors are typically profibrotic and proinflammatory mediators, a novel treatment strategy of CKD could be inhibiting the related signaling, thus suppressing SASP expression. Potential novel agents already exist for this, including anti-fibrotic agents (e.g., TGF-β inhibitor and pirfenidone) and anti-inflammatory agents (e.g., the anti-TNF-α monoclonal antibody and infliximab)
[91][152].