Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Michael Hirth and Version 2 by Jessie Wu.
Pancreatic ductal adenocarcinoma (PDAC) represents one of the most aggressive solid tumors with a dismal prognosis and an increasing incidence. At the time of diagnosis, more than 85% of patients are in an unresectable stage.
- pancreatic ductal adenocarcinoma
- microenvironment
- neuron
- crosstalk
1. Clinical Relevance of Cancer–Neuronal Crosstalk
Besides its aggressive behavior and poor response to treatment, another major feature of pancreatic ductal adenocarcinoma (PDAC) is perineural invasion (PNI) is PNI, which is defined as cancer cells surrounding at least 33% of the epineurial, perineural, and endoneurial space of the nerve sheath. PNI is present in virtually all patients [1][2][10,17]. This is a significant difference from other solid tumors in which PNI is less common [3][18]. PNI can be quantified using scoring systems and is primarily based on its extent and frequency (Figure 1). Interestingly, in PDAC, a significant positive correlation was found between the extent of PNI and patient survival [3][4][5][18,19,20]. PNI is an independent risk factor for the development of R1 resection and tumor recurrence [1][4][10,19] and an important predictor of metastatic spread along the neuronal compartment [2][6][8,17]. For instance, in a retrospective study, Takahashi et al. showed how micrometastases can be frequently found in the neuronal compartment of healthy pancreatic sections of PDAC-bearing patients [2][17]. This event, termed ‘intrapancreatic extratumoral perineural invasion (NEX) phenomenon’, was found in more than 50% of patients undergoing curative surgery. The occurrence of the NEX phenomenon was positively correlated with NI, whilst overall survival was negatively correlated with NEX, as NEX+ patients had a significantly worse survival compared to NEX- (350 vs. 1042 days). All patients who survived PDAC without tumor recurrence were NEX- [2][17].
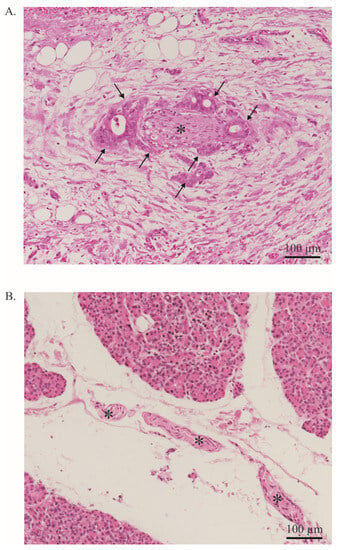
Figure 1. Neural invasion. (A). PDAC cells (arrow) infiltrate a nerve (asterisk). More than 33% of the circumference of the nerve is affected (perineural invasion). Stained with Hematoxylin and Eosin. Image at 20× magnification. (B). Healthy pancreas with a nerve bundle (asterisk). Stained with Hematoxylin and Eosin. The entire tissue shown represents the exocrine part of the pancreas. Endocrine parts are not shown. Image at 20× magnification.
In addition, PNI also has an impact on neuronal architecture. Specifically, extensive nerve fiber hypertrophy and elongation or sprouting of nerve fibers have been usually correlated with PNI occurrence [7][8][21,22]. Nerve fiber hypertrophy appears to be a major contributor to the development of cancer-associated pain in PDAC. Approximately 80% of patients develop cancer-associated pain during the progression of the disease [7][8][21,22]. Even with modern analgesic therapies, cancer-associated pain cannot always be controlled, thus prompting the quest for novel therapeutic targets. Interestingly, two retrospective studies showed that neoadjuvant therapy significantly reduced the rate of PNI [9][10][23,24] As the role of neoadjuvant therapy is currently not well established, this is an interesting observation [11][25]. For instance, Barbier et al. showed that neoadjuvant chemoradiation significantly reduced the rate of PNI from 93 to 43% [9][23]. However, neoadjuvant chemoradiation also prevented more than half of patients from receiving resection due to cancer progression. Consequently, it is unsurprising that neoadjuvant chemoradiation did not improve overall survival compared to direct resection [11][25]. Whether neoadjuvant therapy has a positive impact on patient survival or on the development of cancer-associated pain, e.g., as part of an extended multimodality therapy concept, remains to be elucidated.
2. Molecular Mechanisms of Cancer–Neuronal Crosstalk and Targeted Therapies
2.1. Neurons Are Attracted to Pancreatic Ductal AdenocarcinomaAC Cells
Many mediators are involved in this close cancer–neuronal bidirectional crosstalk. However, our understanding of this complex interaction is still very preliminary. Currently, it is known that PDAC cells secrete mediators that promote nerve invasion into tumor tissue [12][15]. Cancer cells activate physiological mechanisms which in homeostasis are necessary to provide organ innervations and nerve regeneration. Among the identified mediators, neurotrophins, particularly nerve growth factor (NGF), have been best studied (Figure 24A) [12][13][15,78]. During embryonic development, tissues that need to be innervated secrete NGF. Along the NGF gradient, neurons eventually innervate the target organ. In a healthy pancreas, NGF is barely detectable. However, as early as the PanIN stage, the amount of NGF doubled in PDAC cells. In the PDAC stage, the amount of NGF in tumor cells can increase up to seven-fold. Overexpression and secretion of NGF attracts neurons leading to neurite outgrowth into tumor tissue. In human tissue, a clear correlation has been reported between NGF overexpression and an increased extent of NI. Thus, it is not surprising that NGF concentration in tumor tissue is associated with an increased metastasis rate and increased probability of R1 resection. Microdissection studies demonstrated that both PDAC cells and nerves produce NGF and both express the corresponding receptors, so that a reciprocal interaction may be established over the time course of tumor disease [13][78]. NGF binds to two receptors: the high-specificity trkA receptor and the low-specificity p75NTR. Upon binding of NGF to the high-specificity receptor trkA, activation of MEK and MAPK pathways occurs, which promotes proliferation and suppresses apoptosis [14][79]. Binding to the low-affinity p75NTR inhibits proliferation and induces apoptosis [14][79]. In this regard, patients with a high trkA receptor have significantly reduced survival. Patients with high p75NTR expression showed significantly prolonged survival. Interestingly, NGF receptor expression was shown to be one of the most important predictive parameters in PDAC in a multivariable model [15][35]. The crucial role of NGF in this bidirectional crosstalk in PDAC has also been demonstrated in preclinical models. For instance, in vitro, knockdown of NGF or its receptors trkA and p75NTR, respectively, could limit the proliferation and migration of PDAC cells (Figure 2A). Also, inhibition of the NGF—trkA axis resulted in decreased migration of Mia PaCa2 cells toward DRG neurons and decreased neurite outgrowth [1][10]. Similarly, Lei et al. tested the following hypothesis using a robust siRNA (gold nanocluster-associated delivery of siRNA of NGF; GNC-siRNA) for NGF knockdown, which was used both in vitro and in vivo [16][54]. Application of GNC-siRNA reduced proliferation of Panc1 cells and inhibited migration of these tumor cells in a migration chamber. When Panc1 cells were co-cultured with DRG neurons, neurite outgrowth was directed towards Panc1 cells. Once Panc1 cells were pretreated with GNC-siRNA, the extent of neurite outgrowth decreased compared with untreated Panc1 cells. GNC-siRNA was also examined in vivo. Three PDAC mouse models were used (subcutaneous model, orthotopic model and patient-derived xenograft). After NGF knockdown in tumor cells, the amount of neurite outgrowth into tumor tissue was significantly reduced and tumor growth was reduced by approximately 50%. Then, depending on the tumor model, reduction of the metastatic rate varied from a discrete reduction to the virtually complete absence of distant metastasis. Thus, inhibition of the NGF axis represents a promising therapeutic option [16][54]. Human anti-NGF antibodies are already available and used in clinical trials. As NGF also plays an important role in the development and maintenance of pain in chronic inflammation, NGF antibodies have so far been used mainly in clinical trials on painful osteoarthritis [17][80]. The side effects mainly observed include headache, paresthesias and hypoesthesias [17][18][80,81]. Compared to aggressive chemotherapies, these side effects are considered acceptable. To date, no clinical trial for use in pancreatic cancer has been initiated, but this may soon follow due to the promising preclinical data. Specifically, the biweekly application of anti-NGF antibodies had favorable effects in LSL-Kras+/G12D; LSL-Trp53+/R172H; Pdx1-Cre (KPC) animals [19][55]. The development of PDAC was suppressed, the rate of PNI was reduced by 40% and the macrometastases were not detectable (vs. 30% in sham-treated KPC animals). Inhibition of the TrkA receptor also appears to be effective, although human trk inhibitors can cross the blood–brain barrier and thus the side effects to be expected might be more critical [19][55]. Nevertheless, first clinical studies in solid tumors, including pancreatic cancer, have been initiated (Table 1).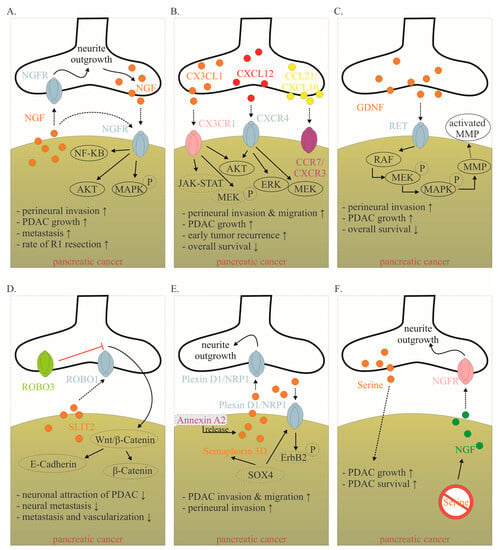
Figure 2. Mediators involved in the cancer–neuronal crosstalk. (A–F). Several mediators are involved in the close cancer–neuronal crosstalk, including NGF, chemokines (e.g., CX3CL1, CXCL10, CCL21), GDNF, SLIT2, Semaphorin 3D and Serine. Solid, black lines with an arrow symbolize activation, whereas red lines with a bar symbolize inhibition. Some signaling mechanisms are only described in non-PDAC cancers. These are labeled with a dotted line. Their role in PDAC has to be confirmed in the future. The effects of interaction (↑ increase; ↓ decrease) are listed below for each mediator. Data collected from [6][12][13][20][21][22][23][24][25][26][27][8,15,78,82,83,84,85,86,87,88,89]. Abbreviations: C-X3-C motif ligand 1 (CX3CL1); CX3C motif chemokine receptor 1 (CX3CR1); C-X-C motif chemokine 10 (CXCL10); C-X-C Motif Chemokine Receptor 3 (CXCR3); C-C Motif) Ligand 21 (CCL21); C-C chemokine receptor type 7 (CCR7); glial-cell-derived neurotrophic factor family of ligands (GDNF); nerve growth factor (NGF); nerve growth factor receptor (NGFR); Pancreatic ductal adenocarcinoma (PDAC); rearranged during transfection (RET); roundabout receptors (Robo); Slit glycoproteins (Slit). Red symbol crossing out Serine means Serine deprivation.
Table 1.
Selected clinical and preclinical trials targeting the cancer–neuronal crosstalk.
| Drug | Target | Current Status | Effect | Reference |
|---|---|---|---|---|
| Anti-NGF antibody or siRNA | NGF | Mouse model | Reduced neural outgrowth, reduced cancer growth, reduced metastasis | [16][19][54,55] |
| Trk-Inhibitor | Trk (unselective) or trkA (selective) | Phase I–II | NA | NCT03556228, NCT05046847, NCT02097810, NCT02568267, NCT04879121 |
| Plerixafor | CXCR4 antagonist | Phase I–II | NA | NCT03277209, NCT02179970, NCT04177810 |
| miR-383 | ROBO3 | Mouse model | Reduced cancer growth and metastasis | [33][56] |
| Minnelide | Several effects, including Inhibition des NF-κB | Phase I–II | NA | NCT03117920, NCT05557851, NCT03129139, NCT04896073 |
| Propranolol | Beta adrenergic receptor | Phase II | NA | NCT03838029, NCT05451043 |
| Celiac ganglion ablation | Denervation | Phase II–III | Reduction in cancer-associated pain, but no significant prolonged survival | [34][57] |
| Splanchnicectomy | Denervation | Phase III | Reduction in cancer-associated pain, prolonged survival only in presence of cancer-associated pain before intervention | [35][58] |
| Bethanechol | Muscarinic receptor M1 | Phase I–II | NA | NCT03572283, NCT05241249 |
Abbreviation: NA (not available; e.g., because the researchtudy is still ongoing or researchstudy results are not yet available).