Oxidative stress is a well-known hallmark of Antiphospholipid Antibody Syndrome (APS), a systemic autoimmune disease characterized by arterial and venous thrombosis and/or pregnancy morbidity. Oxidative stress may affect various signaling pathways and biological processes, promoting dysfunctional immune responses and inflammation, inducing apoptosis, deregulating autophagy and impairing mitochondrial function. The chronic oxidative stress and the dysregulation of the immune system leads to the loss of tolerance, which drives autoantibody production and inflammation with the development of endothelial dysfunction. In particular, anti-phospholipid antibodies (aPL), which target phospholipids and/or phospholipid binding proteins, mainly β-glycoprotein I (β-GPI), play a functional role in the cell signal transduction pathway(s), thus contributing to oxidative stress and thrombotic events. An oxidation–antioxidant imbalance may be detected in the blood of patients with APS as a reflection of disease progression.
- oxidative stress
- antiphospholipid autoantibody
- food supplements
1. Introduction
2. aPL as a Trigger for a Pro-Oxidative State in APS Patients
The role of oxidative stress in the pathogenesis of APS has been widely demonstrated by experimental and clinical studies. It is considered a “second hit” contributing to the induction of the procoagulant phenotype and to the occurrence of thrombosis [6,7,24][6][7][18]. Reactive oxygen species are responsible for lipid peroxidation and the formation of oxidized low-density lipoproteins (oxLDL). oxLDL interact with β2GPI, thus forming complexes with proatherogenic and immunogenic properties. oxLDL/β2GPI complexes can localize inside the intima of the arterial wall and become a target of autoantibodies, thus promoting pro-inflammatory and pro-oxidant pathways and immune cell activation that contribute to the promotion of endothelial dysfunction and thrombotic events [24,25,26][18][19][20]. Patients with primary APS have increased levels of circulating oxidative stress-related markers when compared to healthy subjects. Of interest, APS patients with triple positivity for aPL, i.e., positive for LAC, aCL and aβ2GPI antibodies, show significantly higher levels of these markers than patients with single or double aPL positivity [27][21]. Different mechanisms have been identified as responsible for the induction of oxidative stress in APS. In particular, aPL antibodies have been indicated as a trigger for an increased oxidative status. A previous study by Perez-Sanchez et al. demonstrated that monocytes and neutrophils obtained from APS patients had increased levels of peroxide, NRF2 and antioxidant enzymatic activity, decreased levels of intracellular glutathione and altered mitochondrial membrane potential [28][22]. Of note, aCL antibodies levels positively correlated with the percentage of cells showing depolarized mitochondria and independently predicted the mitochondrial damage detected in monocytes from patients. An increase in peroxide levels and a decrease in both reduced GSH and nuclear NRF2 protein were observed in monocytes stimulated in vitro with IgG from APS patients, but not with IgG from healthy subjects. The treatment with antibodies from patients also increased the percentage of cells with depolarized mitochondria [28][22]. The preincubation of monocytes with antioxidants counteracted the peroxide levels’ increase and restored reduced GSH levels. Furthermore, the pretreatment of the monocytes with Coenzyme Q10, a component of the mitochondrial respiratory chain with antioxidant and anti-inflammatory properties, inhibited ROS production induced by the treatment with IgG from APS patients, thus indicating the mitochondrial electron transport chain as one of the main factors involved in the oxidative perturbation induced by APS autoantibodies [28][22]. Another mechanism implicated in oxidative stress is related to the interactions between aCL antibodies and circulating antioxidant enzymes, such as the paraoxonase-1 (PON1), an antioxidant enzyme that inhibits LDL oxidation. Patients positive for aCL autoantibodies have been demonstrated to show reduced activity of PON1 [30][23]. A cross-sectional study of 77 women positive for antiphospholipid antibodies and 77 controls confirmed these data demonstrating that PON1 activity was lower in women with aPL compared to the controls, and that they had greater structural vascular alterations, and impaired anti-inflammatory and antioxidant properties [31][24]. Data obtained in vitro demonstrated that aPLs also promote oxidative stress by modulating the activity of the pro-oxidant enzymes of immune cells. In particular, it has been demonstrated that human monoclonal aPLs and IgG fractions from APS patients activate endosomal nicotinamide adenine dinucleotide phosphate (NADPH) oxidase and the generation of superoxide in plasmacytoid dendritic cells and monocytes. This activation determines the increased expression of toll-like receptor (TLR) 7 and TLR8 mRNA in plasmacytoid dendritic cells and in monocytes, respectively, and promotes inflammation, which is closely linked to pro-oxidant pathways [35][25]. All these observations sustain the main role of autoantibodies in the induction of a pro-oxidant status in APS patients which, in turn, acts to promote atherothrombosis (Figure 1).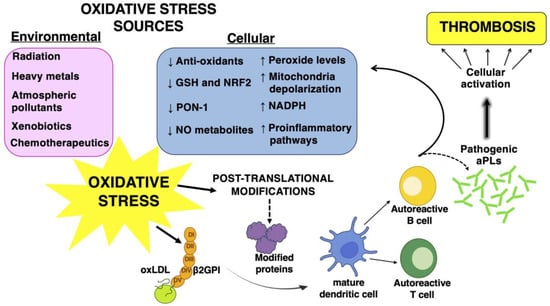
3. Altered Oxidant–Antioxidant Balance in APS Patients: New Biomarkers for Thrombosis Risk Evaluation
Low-to-moderate levels of ROS have important biological functions, as they contribute to the regulation of signal transduction and normal physiological processes. Endogenous antioxidant defense mechanisms play a role in maintaining redox homeostasis [40][26]. Oxidative stress occurs when there is an imbalance between ROS production and cellular antioxidant defenses, and the overproduction of ROS leads to biomolecules damage, particularly in lipids, proteins and DNA [40][26]. Experimental and clinical studies have demonstrated an alteration of the oxidant–antioxidant balance in APS and an increase in oxidative stress-related molecules has been observed in patients with APS. Severe combined immunodeficiency (SCID) mice producing aCL and aβ2GPI had reduced levels of NO and PON1 activity, as well as increased levels of superoxide and peroxynitrite [32][27].
Similar results were obtained in clinical studies. In particular, the presence of high levels of prostaglandin F2-isoprostanes have been detected in plasma from APS patients. These compounds derive from ROS-mediated arachidonic acid oxidation. As F2-isoprostanes are related to lipid peroxidation, they represent an endogenous marker of oxidative stress and have the ability to predict cardiovascular events [42][28]. A cross-sectional study demonstrated that aPL-positive patients had higher levels of isoprostanes, accompanied by higher levels of monocyte Tissue Factor expression, when compared with aPL-negative subjects [43][29].
As oxidative stress has a pivotal role in the pathogenic mechanisms of APS, particularly those related to atherothrombosis, the determination of circulating oxidative stress biomarkers could be useful for the risk assessment of vascular complications in patients with APS. A clinical study conducted in 140 patients with primary and secondary APS aimed to evaluate the relationship between oxidative stress and endothelial damage as a risk factor of thrombosis revealed altered levels of different oxidative stress markers in patients compared to controls. Lipid hydroperoxydes levels were predictive for the impaired flow-mediated dilation of the brachial artery, thus suggesting their possible role as markers of endothelial damage [44][30].
4. The Role of Oxidative Stress in the Post-Translational Modifications of Antigens Associated with APS
The production of ROS at physiological levels cooperates with the resolution of inflammation and the maintenance of homeostasis in tissues [45][31]. An overproduction of ROS and/or a deficiency of the antioxidant machinery can cause a biochemical imbalance and consequent tissue damage [46][32]. The pathogenic roles of oxidative stress and inflammation in APS are also related to their ability to effect protein structural modifications. Pro-oxidant conditions may cause conformational changes in protein structures by promoting post-translational modifications (PTMs) [24][18] (Figure 1). Among PTMs, some can be directly affected by tissue ROS, by inflammatory microenvironments and/or by environmental factors; others, such as acetylation, glycosylation, phosphorylation and citrullination, can be influenced by more indirect downstream pathways affected by ROS [47][33]. PTMs are reversible or irreversible chemical reactions, mainly catalyzed by enzymes that occur in specific amino acids of a certain amount of proteins after their biosynthesis. These processes have a significant impact on the structure and function of proteins; indeed, these modifications play a key role in regulating the folding of proteins, their targeting to specific subcellular compartments, their interaction with ligands or other proteins and, eventually, their immunogenic properties [48,49][34][35]. PTMs can influence cellular processes and consequently, their dysregulation is related to the etiopathogenesis of numerous diseases, in particular, a variety of autoimmune responses depend on PTMs of self-proteins representing a potential trigger of several autoimmune diseases [45][31]. PTMs have been proposed as a link between the environmental factors and oxidative stress inflammation. Environmental factors (smoking, air pollutants, obesity and infections) are associated with PTMs in proteins, and it has been reported as a clear risk factor for breaking tolerance to multiple autoantigens [46,50][32][36]. The known PTMs in APS mainly involve β2GPI. β2GPI is a plasma glycoprotein that consists of five domains rich in cysteines, the V domain is critical for the binding of β2GPI to anionic phospholipids [47][33]. β2GPI plays an important role in the regulation of the blood coagulation system. It is involved in several biological processes, as well as coagulation, fibrinolysis, angiogenesis, thrombosis, autoimmune disease and pregnancy complications. β2GPI may be subjected to PTMs, including oxidation, carbamylation, glycosylation, phosphorylation and acetylation that can have a significant impact on the structure, stability and function of β2GPI in the adoption of an open configuration of the protein that may expose the cryptic epitope and facilitate autoantibody binding. The PTMs directly influence the function of β2GPI and contribute to an increase of its immunogenicity [46,51][32][37]. In particular, PTMs of cysteines include the addition of oxygen. The oxidative/reductive conversion of β2GPI may contribute to the regulation of its biological activity. One of the most functional groups in various proteins is the free sulfhydryl group (SH) contained in the amino acid cysteine. Indeed, the majority of circulating β2GPI is present in a form containing unpaired cysteines (free thiols), which constitutes the reduced form of β2GPI. Sulfhydryl modifications can alter the function of proteins containing cysteines within their catalytic centers or at the protein–protein interaction interface. In an oxidative stress condition, an overproduction of ROS quickly reacts with cysteine residues, especially redox active cysteines, to form reversible or irreversible oxidized forms. Oxidative stress, together with inflammation, is also involved in nonenzymatic glycosylation (glycation of lysine residues) which is a non-enzymatic process that leads to the formation of early, intermediate and advanced glycation end products; these products can modify the structure and function of self-molecules. Elevated levels of antibodies to glucose-modified β2GPI were reported in APS patients. Several factors can modulate this process [51][37]. Another important PTM, related to inflammation and oxidative stress, is carbamylation, a non-enzymatic reaction of cyanate with the primary amine of lysine residues from proteins that generate homocitrulline [23][17]. Particular conditions, such as uremia, inflammation and cigarette smoking, can increase cyanate levels, which are responsible for the formation of these modified proteins [53][38]. While carbamylation has been related to protein aging and other pathological conditions, recent studies have identified carbamylated IgG proteins in patients with rheumatoid arthritis and this suggests that the presence of these antibodies may be useful to predict higher disease activity and may be related to inflammatory biomarkers [23,54][17][39]. Carbamylation occurs in an inflammatory milieu also by neutrophil infiltration and/or in a neutrophil-extracellular traps (NET)/NET-like release [55][40]. Indeed, during inflammation, polymorphonuclear neutrophils and macrophages can release myeloperoxidase (MPO), an enzyme that is stored in azurophilic granules. MPO contributes to the conversion of thiocyanate (derived from food or smoking) as a substrate with H2O2 to cyanate, thereby further increasing carbamylation [53][38]. In APS patients, inflammation, NETosis and oxidative stress may represent the main mechanisms able to induce β2GPI carbamylation [46][32]. Although β2GPI is the most well-known protein in APS, several other proteins were described as antigenic targets and found to play a pathogenetic role in the syndrome. In particular, vimentin contains arginine residue that can undergo PTM to citrulline, catalyzed by the enzyme peptidyl arginine deiminase (PAD) which is active in different cell processes such as in NETosis.5. The Role of Oxidative Stress in the Activation of Multiple Signaling Pathways in Autoimmune Diseases
Oxidative stress is the consequence of the balance alteration between ROS production and a defective detoxification, leading to the alteration of various signaling pathways and multiple biological processes through modifying proteins. These changes in the cellular functions may promote a dysfunctional immune response, inflammation, the induction of apoptosis, deregulation of autophagy, impairment of mitochondrial functions and many other mechanisms. Oxidative imbalance plays a role in the breakdown of immunological tolerance contributing to pathogenesis, pathological progression and the exacerbation of the symptoms in several autoimmune diseases [59,60][41][42] (Figure 1). One of the mechanisms by which oxidative stress can trigger an inflammatory response in the pathogenesis of autoimmune diseases concerns the activation of the enzyme poly (ADP-ribose) polymerase-1 (PARP-1) [61][43]. PARP-1 belongs to enzymes involved in DNA damage sensing and repair and it activates following extensive DNA damage caused by excessive ROS formation [62][44]. PARP-1, through NF-κB activation, may induce the production of inflammatory (TNFα, IL1β and others) and effector T cell cytokines (IL4, IL5), as well as of inflammatory mediators such as metalloproteinases (MMP9), inducible nitric-oxide synthase (iNOS), several chemokines, prostaglandins (PGE2) and alarmins (HMGB1) [63,64,65][45][46][47]. Metabolic cues such as oxidative stress trigger, in most cells, the redox-dependent activation of a mammalian target of rapamycin (mTOR), using a process that involves the cysteine oxidation of Rheb78 and raptor (regulatory-associated protein of mTOR) [68,69][48][49]. Regarding this, oxidative stress plays a role as a regulatory checkpoint in the pathogenesis of systemic lupus erythematosus (SLE) and other autoimmune diseases, where the activation of mTORC1 has been described as a central pathway. In fact, the use of N-acetylcysteine (NAC), an amino acid precursor of glutathione treatment, also reversed the prominent activation of mTORC1 in double-negative T cells [73,74,75][50][51][52]. Oxidatively modified proteins, which act as neoantigens, can trigger pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) of the immune system that represent an important link between some environmental factors such as oxidative stress and autoimmune diseases [76][53]. A condition of oxidative stress can also activate heat shock factors (HSF) and induce a subsequent biosynthesis of heat shock proteins (HSPs), stress-induced cellular molecules that cooperate with the antioxidant system to inhibit or neutralize the cellular effects of ROS [78][54]. Moreover, HSPs are involved in regulating ERK1/2 in the MEK-ERK (mitogen-activated protein kinase) pathway, as well as interacting with Akt, in order to protect cells from apoptosis by the formation of the Akt-HSP complex [79,80][55][56].6. The Interplay among Oxidative Stress, Inflammation and NRF2 Pathways in APS
APS is characterized by uncontrolled inflammation and the over-production of ROS. Increasing evidence supports a link between OS and inflammation in autoimmune diseases [48][34]. Kadl and colleagues showed, in TLR2-deficient mice treated with oxidized phospholipids, how an accumulation of oxidative tissue damage creates a microenvironment that causes “sterile” TLR2-dependent chronic inflammation [85][57]. Recently, Wang G et al., using lupus-prone MRL+/+ mice, demonstrated that trichloroethene (TCE) exposure accelerated an autoimmune response by inducing the dysregulation of TLR signaling and the concomitant impairment of NRF2 and its target gene HO-1. Antioxidant supplementation clearly ameliorated TCE-induced reduction in both NRF2 and HO-1 [86][58]. In addition, the NRF2/ARE signaling pathway may be considered the primary pathway for intracellular redox balance and the molecules that it induces exert antioxidant and anti-inflammatory effects by upregulating various cytoprotective enzymes and proteins. NRF2 is physiologically localized in the cytoplasm and anchored to the intracytoplasmic actin cytoskeleton in the form of the Keap1–NRF2 complex. E3 ubiquitin ligase promotes the ubiquitin-mediated degradation of NRF2 and the maintenance of a basal steady-state level of NRF2 activity. Otherwise, under oxidative or electrophilic stress conditions, NRF2 dissociates from Keap1 and translocates to the nucleus forming a heterodimer with the small musculoaponeurotic fibrosarcoma (sMAF) proteins and binds to an Antioxidant Response Element (ARE) in the promoter region of cytoprotective genes [87][59]. Interest in the cytoprotective role of NRF2 is growing as a potential drug target in APS, in which OS and inflammation underlie the disease’s pathogenesis. In patients with APS, the NRF2 activation was repressed in monocytes, thus suggesting a defect in the NRF2 signaling pathway [28][22]. In an animal study, mice lacking NRF2 developed several characteristics of the autoimmune-mediated lesions, similar to those of human SLE, the spontaneous development of autoantibodies, increased T-cell proliferation, multi-tissue inflammatory lesions, along with glomerulonephritis, the intravascular deposition of immunoglobulin complexes and premature death [93][60].References
- Lim, W. Antiphospholipid syndrome. Hematol. Am. Soc. Hematol. Educ. Progr. 2013, 2013, 675–680.
- Hughes, G.R. The anticardiolipin syndrome. Clin. Exp. Rheumatol. 1985, 3, 285–286.
- Hughes, G.R.; Harris, N.N.; Gharavi, A.E. The anticardiolipin syndrome. J. Rheumatol. 1986, 13, 486–489.
- Pastori, D.; Ames, P.R.J.; Triggiani, M.; Ciampa, A.; Cammisotto, V.; Carnevale, R.; Pignatelli, P.; Bucci, T. Antiphospholipid Antibodies and Heart Failure with Preserved Ejection Fraction. The Multicenter ATHERO-APS Study. J. Clin. Med. 2021, 10, 3180.
- Abreu, M.M.; Danowski, A.; Wahl, D.G.; Amigo, M.C.; Tektonidou, M.; Pacheco, M.S.; Fleming, N.; Domingues, V.; Sciascia, S.; Lyra, J.O.; et al. The relevance of “non-criteria” clinical manifestations of antiphospholipid syndrome: 14th International Congress on Antiphospholipid Antibodies Technical Task Force Report on Antiphospholipid Syndrome Clinical Features. Autoimmun. Rev. 2015, 14, 401–414.
- Shoenfeld, Y.; Meroni, P.L.; Toubi, E. Antiphospholipid syndrome and systemic lupus erythematosus: Are they separate entities or just clinical presentations on the same scale? Curr. Opin. Rheumatol. 2009, 21, 495–500.
- Meroni, P.L.; Borghi, M.O.; Raschi, E.; Tedesco, F. Pathogenesis of antiphospholipid syndrome: Understanding the antibodies. Nat. Rev. Rheumatol. 2011, 7, 330–339.
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13.
- Sandalio, L.M.; Rodríguez-Serrano, M.; Romero-Puertas, M.C.; del Río Luis, A. Role of peroxisomes as a source of reactive oxygen species (ROS) signaling molecules. Subcell. Biochem. 2013, 69, 231–255.
- Cao, S.S.; Kaufman, R.J. Endoplasmic reticulum stress and oxidative stress in cell fate decision and human disease. Antioxid. Redox Signal. 2014, 21, 396–413.
- Naidoo, K.; Birch-Machin, M.A. Oxidative stress and ageing: The influence of environmental pollution, sunlight and diet on skin. Cosmetics 2017, 4, 4.
- Murphy, M.P.; Holmgren, A.; Larsson, N.G.; Halliwell, B.; Chang, C.J.; Kalyanaraman, B.; Rhee, S.G.; Thornalley, P.J.; Partridge, L.; Gems, D.; et al. Unraveling the biological roles of reactive oxygen species. Cell Metab. 2011, 13, 361–366.
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535.
- Rybicka, J.M.; Balce, D.R.; Khan, M.F.; Krohn, R.M.; Yates, R.M. NADPH oxidase activity controls phagosomal proteolysis in macrophages through modulation of the lumenal redox environment of phagosomes. Proc. Natl. Acad. Sci. USA 2010, 107, 10496–10501.
- Yang, H.C.; Cheng, M.L.; Ho, H.Y.; Tsun-Yee Chiu, D. The microbicidal and cytoregulatory roles of NADPH oxidases. Microbes Infect. 2011, 13, 109–120.
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. Biophys. Acta 2016, 1863, 2977–2992.
- Buttari, B.; Profumo, E.; Capozzi, A.; Saso, L.; Sorice, M.; Riganò, R. Post-translational modifications of proteins in antiphospholipid antibody syndrome. Crit. Rev. Clin. Lab. Sci. 2019, 56, 511–525.
- Nocella, C.; Bartimoccia, S.; Cammisotto, V.; D’amico, A.; Pastori, D.; Frati, G.; Sciarretta, S.; Rosa, P.; Felici, C.; Riggio, O.; et al. Oxidative Stress in the Pathogenesis of Antiphospholipid Syndrome: Implications for the Atherothrombotic Process. Antioxidants 2021, 10, 1790.
- Matsuura, E.; Kobayashi, K.; Hurley, B.L.; Lopez, L.R. Atherogenic oxidized low-density lipoprotein/beta2-glycoprotein I (oxLDL/beta2GPI) complexes in patients with systemic lupus erythematosus and antiphospholipid syndrome. Lupus 2006, 15, 478–483.
- Zhang, G.; Cai, Q.; Zhou, H.; He, C.; Chen, Y.; Zhang, P.; Wang, T.; Xu, L.; Yan, J. OxLDL/β2GPI/anti-β2GPI Ab complex induces inflammatory activation via the TLR4/NF-κB pathway in HUVECs. Mol. Med. Rep. 2021, 23, 148.
- Sciascia, S.; Roccatello, D.; Bertero, M.T.; Di Simone, D.; Cosseddu, D.; Vaccarino, A.; Bazzan, M.; Rossi, D.; Garcia-Fernandez, C.; Ceberio, L.; et al. 8-Isoprostane, prostaglandin E2, C-reactive protein and serum amyloid A as markers of inflammation and oxidative stress in antiphospholipid syndrome: A pilot study. Inflamm. Res. 2012, 61, 809–816.
- Perez-Sanchez, C.; Ruiz-Limon, P.; Aguirre, M.A.; Bertolaccini, M.L.; Khamashta, M.A.; Rodriguez-Ariza, A.; Segui, P.; Collantes-Estevez, E.; Barbarroja, N.; Khraiwesh, H.; et al. Mitochondrial dysfunction in antiphospholipid syndrome: Implications in the pathogenesis of the disease and effects of coenzyme Q(10) treatment. Blood 2012, 119, 5859–5870.
- Lambert, M.; Boullier, A.; Hachulla, E.; Fruchart, J.C.; Teissier, E.; Hatron, P.Y.; Duriez, P. Paraoxonase activity is dramatically decreased in patients positive for anticardiolipin antibodies. Lupus 2000, 9, 299–300.
- Charakida, M.; Besler, C.; Batuca, J.R.; Sangle, S.; Marques, S.; Sousa, M.; Wang, G.; Tousoulis, D.; Delgado Alves, J.; Loukogeorgakis, S.P.; et al. Vascular abnormalities, paraoxonase activity, and dysfunctional HDL in primary antiphospholipid syndrome. JAMA 2009, 302, 1210–1217.
- Prinz, N.; Clemens, N.; Strand, D.; Pütz, I.; Lorenz, M.; Daiber, A.; Stein, P.; Degreif, A.; Radsak, M.; Schild, H.; et al. Antiphospholipid antibodies induce translocation of TLR7 and TLR8 to the endosome in human monocytes and plasmacytoid dendritic cells. Blood 2011, 118, 2322–2332.
- Milne, G.L.; Yin, H.; Hardy, K.D.; Davies, S.S.; Roberts, L.J. Isoprostane generation and function. Chem. Rev. 2011, 111, 5973–5996.
- Delgado Alves, J.; Mason, L.J.; Ames, P.R.J.; Chen, P.P.; Rauch, J.; Levine, J.S.; Subang, R.; Isenberg, D.A. Antiphospholipid antibodies are associated with enhanced oxidative stress, decreased plasma nitric oxide and paraoxonase activity in an experimental mouse model. Rheumatology 2005, 44, 1238–1244.
- Pignatelli, P.; Pastori, D.; Carnevale, R.; Farcomeni, A.; Cangemi, R.; Nocella, C.; Bartimoccia, S.; Vicario, T.; Saliola, M.; Lip, G.Y.H.; et al. Serum NOX2 and urinary isoprostanes predict vascular events in patients with atrial fibrillation. Thromb. Haemost. 2015, 113, 617–624.
- Ferro, D.; Saliola, M.; Meroni, P.L.; Valesini, G.; Caroselli, C.; Praticó, D.; Fitzgerald, G.A.; Shoenfeld, Y.; Violi, F. Enhanced monocyte expression of tissue factor by oxidative stress in patients with antiphospholipid antibodies: Effect of antioxidant treatment. J. Thromb. Haemost. 2003, 1, 523–531.
- Stanisavljevic, N.; Stojanovich, L.; Marisavljevic, D.; Djokovic, A.; Dopsaj, V.; Kotur-Stevuljevic, J.; Martinovic, J.; Memon, L.; Radovanovic, S.; Todic, B.; et al. Lipid peroxidation as risk factor for endothelial dysfunction in antiphospholipid syndrome patients. Clin. Rheumatol. 2016, 35, 2485–2493.
- Haro, I.; Sanmartí, R.; Gómara, M.J. Implications of Post-Translational Modifications in Autoimmunity with Emphasis on Citrullination, Homocitrullination and Acetylation for the Pathogenesis, Diagnosis and Prognosis of Rheumatoid Arthritis. Int. J. Mol. Sci. 2022, 23, 15803.
- Misasi, R.; Longo, A.; Recalchi, S.; Caissutti, D.; Riitano, G.; Manganelli, V.; Garofalo, T.; Sorice, M.; Capozzi, A. Molecular Mechanisms of “Antiphospholipid Antibodies” and Their Paradoxical Role in the Pathogenesis of “Seronegative APS”. Int. J. Mol. Sci. 2020, 21, 8411.
- Passam, F.H.; Giannakopoulos, B.; Mirarabshahi, P.; Krilis, S.A. Molecular pathophysiology of the antiphospholipid syndrome: The role of oxidative post-translational modification of beta 2 glycoprotein I. J. Thromb. Haemost. 2011, 9 (Suppl. S1), 275–282.
- Carubbi, F.; Alunno, A.; Gerli, R.; Giacomelli, R. Post-Translational Modifications of Proteins: Novel Insights in the Autoimmune Response in Rheumatoid Arthritis. Cells 2019, 8, 657.
- Ramazi, S.; Zahiri, J. Posttranslational modifications in proteins: Resources, tools and prediction methods. Database 2021, 2021, baab012.
- Ebert, T.; Tran, N.; Schurgers, L.; Stenvinkel, P.; Shiels, P.G. Ageing-Oxidative stress, PTMs and disease. Mol. Aspects Med. 2022, 86, 101099.
- Brusch, A. The Significance of Anti-Beta-2-Glycoprotein I Antibodies in Antiphospholipid Syndrome. Antibodies 2016, 5, 16.
- Darrah, E.; Andrade, F. Editorial: Citrullination, and carbamylation, and malondialdehyde-acetaldehyde! Oh my! Entering the forest of autoantigen modifications in rheumatoid arthritis. Arthritis Rheumatol. 2015, 67, 604–608.
- Sorice, M.; Buttari, B.; Capozzi, A.; Profumo, E.; Facchiano, F.; Truglia, S.; Recalchi, S.; Alessandri, C.; Conti, F.; Misasi, R.; et al. Antibodies to age-β2 glycoprotein I in patients with anti-phospholipid antibody syndrome. Clin. Exp. Immunol. 2016, 184, 174–182.
- Capozzi, A.; Truglia, S.; Buttari, B.; Recalchi, S.; Riitano, G.; Manganelli, V.; Mancuso, S.; Alessandri, C.; Longo, A.; Mattei, V.; et al. Carbamylation of β2-glycoprotein I generates new autoantigens for antiphospholipid syndrome: A new tool for diagnosis of “seronegative” patients. Rheumatology 2022, 61, 4187–4197.
- Khan, M.F.; Wang, G. Environmental Agents, Oxidative Stress and Autoimmunity. Curr. Opin. Toxicol. 2018, 7, 22–27.
- Ramani, S.; Pathak, A.; Dalal, V.; Paul, A.; Biswas, S. Oxidative Stress in Autoimmune Diseases: An Under Dealt Malice. Curr. Protein Pept. Sci. 2020, 21, 611–621.
- Wang, G.; Ma, H.; Wang, J.; Khan, M.F. Contribution of poly(ADP-ribose)polymerase-1 activation and apoptosis in trichloroethene-mediated autoimmunity. Toxicol. Appl. Pharmacol. 2019, 362, 28–34.
- Pazzaglia, S.; Pioli, C. Multifaceted Role of PARP-1 in DNA Repair and Inflammation: Pathological and Therapeutic Implications in Cancer and Non-Cancer Diseases. Cells 2019, 9, 41.
- Zerfaoui, M.; Errami, Y.; Naura, A.S.; Suzuki, Y.; Kim, H.; Ju, J.; Liu, T.; Hans, C.P.; Kim, J.G.; Abd Elmageed, Z.Y.; et al. Poly(ADP-ribose) polymerase-1 is a determining factor in Crm1-mediated nuclear export and retention of p65 NF-kappa B upon TLR4 stimulation. J. Immunol. 2010, 185, 1894–1902.
- Ditsworth, D.; Zong, W.X.; Thompson, C.B. Activation of poly(ADP)-ribose polymerase (PARP-1) induces release of the pro-inflammatory mediator HMGB1 from the nucleus. J. Biol. Chem. 2007, 282, 17845–17854.
- Kay, J.; Thadhani, E.; Samson, L.; Engelward, B. Inflammation-induced DNA damage, mutations and cancer. DNA Repair 2019, 83, 102673.
- Yoshida, S.; Hong, S.; Suzuki, T.; Nada, S.; Mannan, A.M.; Wang, J.; Okada, M.; Guan, K.L.; Inoki, K. Redox regulates mammalian target of rapamycin complex 1 (mTORC1) activity by modulating the TSC1/TSC2-Rheb GTPase pathway. J. Biol. Chem. 2011, 286, 32651–32660.
- Perl, A. Activation of mTOR (mechanistic target of rapamycin) in rheumatic diseases. Nat. Rev. Rheumatol. 2016, 12, 169–182.
- Wu, T.; Ye, Y.; Min, S.Y.; Zhu, J.; Khobahy, E.; Zhou, J.; Yan, M.; Hemachandran, S.; Pathak, S.; Zhou, X.J.; et al. Prevention of murine lupus nephritis by targeting multiple signaling axes and oxidative stress using a synthetic triterpenoid. Arthritis Rheumatol. 2014, 66, 3129–3139.
- Lai, Z.W.; Hanczko, R.; Bonilla, E.; Caza, T.N.; Clair, B.; Bartos, A.; Miklossy, G.; Jimah, J.; Doherty, E.; Tily, H.; et al. N-acetylcysteine reduces disease activity by blocking mammalian target of rapamycin in T cells from systemic lupus erythematosus patients: A randomized, double-blind, placebo-controlled trial. Arthritis Rheum. 2012, 64, 2937–2946.
- Tan, M.K.X.; Heng, T.Y.J.; Mak, A. The potential use of metformin, dipyridamole, N-acetylcysteine and statins as adjunctive therapy for systemic lupus erythematosus. Cells 2019, 8, 323.
- Ryan, B.J.; Nissim, A.; Winyard, P.G. Oxidative post-translational modifications and their involvement in the pathogenesis of autoimmune diseases. Redox Biol. 2014, 2, 715–724.
- Szyller, J.; Bil-Lula, I. Heat Shock Proteins in Oxidative Stress and Ischemia/Reperfusion Injury and Benefits from Physical Exercises: A Review to the Current Knowledge. Oxid. Med. Cell. Longev. 2021, 2021, 6678457.
- Ikeyama, S.; Kokkonen, G.; Shack, S.; Wang, X.T.; Holbrook, N.J. Loss in oxidative stress tolerance with aging linked to reduced extracellular signal-regulated kinase and Akt kinase activities. FASEB J. 2002, 16, 114–116.
- Cao, W.; Li, M.; Li, J.; Li, C.; Xu, X.; Gu, W. Geranylgeranylacetone ameliorates lung ischemia/reperfusion injury by HSP70 and thioredoxin redox system: NF-κB pathway involved. Pulm. Pharmacol. Ther. 2015, 32, 109–115.
- Kadl, A.; Sharma, P.R.; Chen, W.; Agrawal, R.; Meher, A.K.; Rudraiah, S.; Grubbs, N.; Sharma, R.; Leitinger, N. Oxidized phospholipid-induced inflammation is mediated by Toll-like receptor 2. Free Radic. Biol. Med. 2011, 51, 1903–1909.
- Wang, G.; Wang, H.; Banerjee, N.; Khan, M.F. Interplay and roles of oxidative stress, toll-like receptor 4 and Nrf2 in trichloroethene-mediated autoimmunity. Toxicol. Appl. Pharmacol. 2020, 408, 115258.
- Baird, L.; Yamamoto, M. The Molecular Mechanisms Regulating the KEAP1-NRF2 Pathway. Mol. Cell. Biol. 2020, 40, e00099-20.
- Ma, Q.; Battelli, L.; Hubbs, A.F. Multiorgan autoimmune inflammation, enhanced lymphoproliferation, and impaired homeostasis of reactive oxygen species in mice lacking the antioxidant-activated transcription factor Nrf2. Am. J. Pathol. 2006, 168, 1960–1974.