Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Myron R. Szewczuk and Version 2 by Catherine Yang.
Angiogenesis is seen as the process that mediates new blood vessel formation and capillaries, an essential process allowing for the exchange of nutrients throughout the body. Angiogenic growth factors (AFs) influence both innate and adaptive immune cell populations within the tumor microenvironment (TME) to create a more tolerogenic milieu. Myeloid regulatory cells such as myeloid-derived suppressor cells (MDSCs), tumor-associated macrophages (TAMs), type 2 natural killer T (NKT) cells, and regulatory T-cells (Tregs) are the primary cells types that contribute to immune escape and immunosuppression within the tumor microenvironment.
- angiogenesis
- anti-angiogenic treatment
- control systems
1. Tumor-Associated Macrophages and Angiogenic Growth Factors
Tumor-associated macrophages differentiate into anti-inflammatory M1 macrophages (M1-TAMS) or tumor-promoting and proinflammatory M2 macrophages (M2-TAMS) [1][2][17,18]. The polarization of TAMs into their respective phenotypes is dependent on local signals provided in the tumor milieu [1][17]. VEGF, directly and indirectly, polarizes monocytes towards an M2 phenotype, contributing to an immunosuppressive tumor microenvironment [3][19]. M2-TAMS express cytokines IL-6, IL-10, CCl-22, and TGF-β, which, while promoting monocyte maturation, block the differentiation of monocytes to dendritic cells and disrupt the activity of cytotoxic T lymphocytes (CTLs) and NK cells [4][5][20,21]. It was previously understood that VEGF induces monocyte recruitment to the tumor site by stimulating endothelial cells to release monocyte chemoattractant protein (MCP-1), increasing the endothelial layer’s permeability to enhance cell migration [6][22]. Monocytes would thus have increased opportunity for direct contact with tumor cells and exposure to the various signals present in the tumor microenvironment, skewing monocytes to differentiate into an M2-TAM phenotype [1][17] (Figure 1).
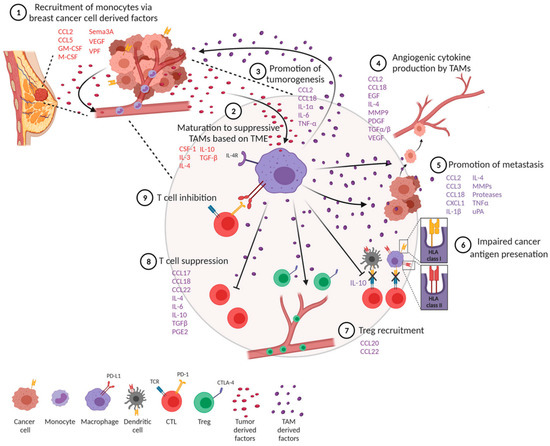
Figure 1. Overview of the role TAM polarization and cytokine/growth factor recruitment plays in breast cancer tumorigenesis, metastasis, and immune evasion. Citation: © 2021 Mehta, Kadel, Townsend, Oliwa and Guerriero. Frontiers in immunology 2021, 12:643,771, https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8102870/ (accessed on 27 July 2023). This is an Open Access article that permits unrestricted non-commercial use, provided the original work is properly cited.
PIGF binding to VEGFR-1 stimulates the recruitment of macrophages into the tumor microenvironment [7][8][9][10][11][23,24,25,26,27]. PIGF is upregulated in many advanced stages of cancer and may also play a role in monocyte recruitment, separate from its stimulation of VEGF secretion. Previously, it was established that factors like VEGF could influence M2-TAMs and promote their polarization towards the M2 phenotype associated with tumor-promoting functions. However, recent data suggest that in the context of M2-TAM polarization, the signaling of PlGF might play a more critical role than VEGF signaling through VEGFR-1. Macrophage polarization and its association with PlGF have been linked to Histidine-rich glycoprotein (HRG), a plasma protein with anti-inflammatory effects [5][21]. By binding to different cells, HRG, produced by monocytes and macrophages, can modulate various functions, including immunity and vascularization. HRG exhibits both anti-angiogenic and pro-angiogenic activity, which is believed to be due to its disruption of the endothelial cell cytoskeleton, inhibiting vessel formation. [5][21]. In contrast, HRG’s pro-angiogenic activity results from its high-affinity binding to thrombospondin-1 (TSP-1), masking TSP-1′s anti-angiogenic epitope [10][26]. The inhibition of angiogenesis by TSP-1 and HRG depends on TSP-1 interacting with CD47, which not only inhibits endothelial cell proliferation but also downstream eNOS/NO/cGMP signaling and VEGFR-2 phosphorylation [12][28]. Importantly, this inhibition of VEGFR-2 phosphorylation does not prevent the binding of VEGF-VEGFR [12][28].
Studies have shown that overexpression of HRG in specific cancer cells leads to slower tumor growth and reduced metastasis in mice, even with persistent TAMs accumulation [13][29]. HRG exposure downregulates M2 markers in TAMs, such as IL10, CCL22, and PlGF, while upregulating M1 markers, such as IL6 and CXCL9 [7][23]. This shift in TAM polarization helps decrease the immunosuppression within the tumor microenvironment by decreasing regulatory T-cell (Treg) recruitment, improving DCs and T-cells function, and promoting infiltration of CD8+ T-cells and NK cells into the tumor stroma [7][23].
HRG’s effect on TAMs polarization appears to be modulated mainly by the downregulation of PlGF. Without host derived PlGF, HRG does not further suppress tumor growth. One study showed that PlGF expressed by non-small cell lung cancer (NSCLC) cells triggers TAM polarization and promotes tumor growth and metastasis [13][29]. In breast cancer (BC) and murine pancreatic ductal adenocarcinoma (PDAC) models implanted in obese mice, targeting PlGF/VEGFR-1 signaling led to a shift in the profile of tumor secreted cytokines and TAMs differentiation towards the M1 phenotype, resulting in reduced tumor progression [14][30]. It was found that plasma PlGF was associated with obesity in PDAC and BC patient samples and that VEGF-A was not, further supporting the role of PlGF in TAM polarization towards the M2 phenotype [14][30]. In a more recent study, Ma et al. [15][31] demonstrated that using metformin (200 mg/kg day) was enough to repolarize M2-TAMs into M1-TAMs and to decrease tumor progression even in the presence of PIGF autocrine signaling. Another study found that an inhibitor of HIF-1α, Lificiguat (YC-1), in triple-negative breast cancer (TNBC), also had similar results in repolarizing TAMs and inhibiting angiogenesis and tumor growth [16][32].
These findings suggest that PlGF, rather than VEGF through VEGFR-1, is a crucial driver of TAM polarization towards the immunosuppressive M2 phenotype [5][21]. This highlights the potential importance of targeting PlGF signaling as a therapeutic strategy to modulate TAM polarization and promote an anti-tumoral immune response within the tumor microenvironment. It is also essential to recognize that many factors, such as FGF, are highly associated with M2 polarization, whose mechanisms are yet to be fully elucidated [17][10]. Further research is required to understand the role of various AFs across tumor heterogeneity.
2. Myeloid-Derived Suppressor Cells (MDSCs) and Angiogenic Growth Factors
M2 polarized macrophages secrete Th2 cytokines, such as IL-10, TGF-β, CCL-22, IL-6, and growth factors, such as VEGF and PIGF, contributing to angiogenic remodeling [1][17]. VEGF, IL-6, CCL-22, and IL-10 are also linked to further stimulation and recruitment of MDSCs to the TME [18][19][33,34]. MDSCs contribute to a more immunosuppressive tumor milieu by secreting anti-inflammatory cytokines such as IL-10 and TGF-β [20][35]. TGF-β contributes to the induction and production of Tregs into the TME, and IL-10 can arrest the production of interferon-γ (IFN-γ) by CD4+ T-cells, which further drives cancer progression and metastasis [21][36]. MDSCs, as an immature and undifferentiated population, can further differentiate into macrophages such as M2-TAMs, and DCs [21][36]. MDSCs, as immature myeloid cells, are well known to depress the T-cell function. Immune checkpoint regulators, such as the programmed-death ligand 1 (PD-L1), are known to be expressed on MDSCs [21][36]. HIF-1α regulates PD-L1 gene expression. Thus, under hypoxic conditions such as the environment within the TME, PD-L1 is overexpressed by MDSCs [22][37]. The programmed death 1 receptor (PD-1) to which PD-L1 binds is expressed by the effector CD4+ and CD8+ T-cells, DCs, and APCs [21][36]. PD-L1 on MDSCs interact with T-cells which express PD-1, resulting in T-cell anergy, where there is diminished cytokine production [21][36]. Additionally, MDSCs carry exosomal cargo rich in cytokines and AFs, which drive cancer progression and metastasis when released into the TME [21][23][36,38]. MDSCs use abundant VEGF in the TME and bind to VEGFR to initiate signaling cascades involving JAK2/STAT3 to produce additional angiogenic molecules [21][36]. Interestingly, it was found that the activation of MDSCs by VEGF resulted in increased immunosuppressive activity relative to non-exposed MDSCs [24][39]. Stimulation of MDSCs by proinflammatory cytokines and VEGF allows the production of VEGF through a STAT-3-mediated pathway. This positive feedback loop recruits further MDSCs to the TME and results in the further release of AFs [21][36].
3. Dendritic Cells, T-Cells, Tregs Signaling, and Angiogenic Growth Factors
Dendritic cells (DCs) are crucial in activating T-cells through antigen presentation. However, various AFs have been found to inhibit dendritic cell maturation and antigen presentation. Recent work has demonstrated an association between high levels of VEGF expression in human cancers, impaired cell function, and a reduced number of cells [25][40]. Recall that VEGF, PIGF, and FGF are involved in the recruitment of cells such as M2-TAMs and MDSC, which can block the differentiation of DC and increase PD-L1 expression within the TME. VEGF upregulates PD-L1 in dendritic cells, inhibiting T-cell expansion and function, and inhibits the function of mature dendritic cells to stimulate T-cells by acting on VEGFR-2 and inhibiting NF-κB activation [26][27][41,42]. It also interferes with the ability of mature dendritic cells to stimulate T-cells through the involvement of VEGFR-2. FGF/FGFR signaling through the JAK/STAT pathway has also been associated with increased expression of PD-L1 through the upregulation of Yes-associated protein (YAP) [17][26][10,41]. YAP is considered to have an oncogenic role, aiding in inhibiting tumor apoptosis and triggering metastasis and therapeutic evasion [28][43]. PIGF is also implicated in the suppression of DCs and inhibition of T-cell proliferation through the binding of PIGF to VEGFR-1 [29][30][44,45]. Moreover, PIGF has been found to upregulate the secretion of anti-inflammatory cytokines such as IL-10 by CD4+ and CD8+ T-cells leading to increased immunosuppression [29][44].
VEGF contributes to CD8+ T-cell exhaustion through VEGFR-2 and activated T-cell nuclear factor-mediated process [26][31][41,46]. CD8+ T-cell exhaustion can occur due to the expression of negative immune checkpoints such as PD-1, CTLA-4, T-cell immunoglobulin mucin receptor 3, and lymphocyte activation gene 3 protein [26][31][41,46]. Furthermore, the induction of FAS-ligand expression on endothelial cells by VEGF establishes a selective immune barrier that can suppress effector T-cell functions and cause apoptosis of CD8+ T-cells [32][47]. VEGF also promotes Treg generation, impeding CD8+ and CD4+ T-cell differentiation within the thymus [26][41]. VEGF can function as a chemoattractant in recruiting FOXP3+ Treg cells, suppressing the anti-tumor response [26][27][41,42]. VEGF signaling through VEGFR-2 also aids in the induction and survival of Tregs within the TME [27][42]. FGF/FGFR signaling has also increased the survival of Tregs through IL-2-mediated STAT5 phosphorylation [17][10].