Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Camila Xu and Version 1 by Razvan Adrian Covache-Busuioc.
An subarachnoid hemorrhage (SAH) is the predominant initial sign of cerebral aneurysms in both adults and kids. In children, the incidence of an SAH varies between 1.9% and 4.6%. The growing detection of SAHs in children can probably be attributed to better diagnostic tools and heightened clinical vigilance.
- intracranial aneurysm
- genetic syndromes
- genome-wide association studies
1. Introduction
About 80% of all non-traumatic subarachnoid hemorrhages (SAHs) are caused by intracranial aneurysms (IAs), commonly referred to as saccular or berry aneurysms. Between 2% and 5% of people have intracranial aneurysms. Of these, 0.7% to 1.9% will experience a rupture leading to subarachnoid hemorrhage [1,2][1][2].
An SAH is the predominant initial sign of cerebral aneurysms in both adults and kids. In children, the incidence of an SAH varies between 1.9% and 4.6%. The growing detection of SAHs in children can probably be attributed to better diagnostic tools and heightened clinical vigilance [3].
However, the occurrence of an IA seems to increase with age [4]. For those over 30 years old, the likelihood of having an IA ranges from 3.6% to 6.5% [5]. Women are more predisposed to aneurysms than men, with a 3:1 ratio in cases of unruptured IAs. About 70–75% of IAs appear individually, whereas 25–30% occur as multiple aneurysms [6,7][6][7].
Among adults, unruptured IAs have a prevalence rate of about 2% to 6%. They generally do not show symptoms and are usually discovered during MRI or CT scans for other neurological reasons or when screening high-risk individuals [8].
The frequency of SAHs due to IAs varies globally. Finland and Japan have the highest rates, between 22.5 and 32 per 100,000 people annually. Globally, this rate stands at 9.1 per 100,000 individuals every year [9]. An SAH strikes early in life and is fatal, contributing to more than 25% of years of life lost for stroke sufferers below 65 years of age [10].
The majority of individuals are unaware that they have an aneurysm until it bursts. Some might experience precursor symptoms like pain around the eye, nerve paralysis in the face, or headaches and neck pain from a minor aneurysm leak, known as a “sentinel bleed” [11].
2. Risk Factors Associated with IAs
Habits and health conditions such as active smoking, a high blood pressure, and excessive alcohol consumption are recognized as separate risk factors that can contribute to the development of an aneurysmal subarachnoid hemorrhage [12]. Additional risk factors include an advancing age and being female, since the yearly occurrence rate for men was determined to be 4.5 per 100,000 with a confidence interval (CI) of 3.1 to 5.8, while for women, it was 7.1 per 100,000 with a CI of 5.4 to 8.7. The risk for women, when compared to men, had a relative value of 1.6 with a CI of 1.1 to 2.3 [13]. Moreover, having a family history of intracranial aneurysms, and the consumption of drugs that stimulate the sympathetic nervous system, commonly known as sympathomimetic drugs, are other risk factors [14]. Genetic conditions can further heighten the risk of an aneurysmal SAH. For instance, those with autosomal dominant polycystic kidney disease or vascular Ehlers-Danlos syndrome (often referred to as type IV EDS) are more susceptible.
Recent research, like the PHASES study, indicates that one’s geographical origin, such as being of Finnish or Japanese descent, can be a strong determinant for aneurysmal rupture. This potentially suggests that genetic predispositions, associated with certain regions or ethnicities, can play a significant role in rupture risk [15]. The 1999 study by the MARS group provided clarity on the importance of screening close relatives of a patient diagnosed with an IA. Their findings underscored the preventive value of screening: for every 149 first-degree relatives screened using Magnetic Resonance Angiography (MRA), one SAH could be averted. To prevent a single SAH-related death, 298 patients would need to be screened [16]. A family history of IA stands out as the most telling risk marker for the condition. To illustrate, family members from a family with at least two patients diagnosed with IA have about a four-fold increased risk of having an IA, as shown by MRA screening, compared to the general public [4].
Although several inheritable conditions, including autosomal dominant polycystic kidney disease, neurofibromatosis type I, Marfan syndrome, multiple endocrine neoplasia type I, pseudoxanthoma elasticum, hereditary hemorrhagic telangiectasia, and Ehlers-Danlos syndrome types II and IV, have links to the formation of IAs, they represent less than 1% of all IAs in the general population. As such, these genetic syndromes cannot account for the majority of familial clusters of IA cases. However, when family histories are thoroughly researched, familial occurrences of IA seem to be more frequent than initially thought.
2.1. Genetic Syndromes with Increased Intracranial Aneurysm Incidence
Autosomal Dominant Polycystic Kidney Disease (ADPKD): Through comprehensive literary reviews, it has been established that individuals with ADPKD have a roughly 11% prevalence rate of intracranial aneurysms [18][17]. ADPKD’s genetic origin lies in the loss-of-function of either the polycystin 1 (PKD1) or polycystin 2 (PKD2) genes. This primarily results in the development of cysts in the kidneys, which can impair renal function and eventually progress to kidney failure [19][18]. Beyond the kidneys, ADPKD can impact other organs, particularly the liver. A common comorbidity among ADPKD patients is hypertension, which is a known risk factor not just for IA, but for other cardiovascular diseases as well [20][19]. The life expectancy for those with ADPKD is notably reduced, with PKD1 and PKD2 patients in Europe averaging lifespans of 53 and 69 years, respectively [21][20].
Microcephalic Osteodysplastic Primordial Dwarfism Type II (MOPDII): This is the most prevalent variant of microcephalic primordial dwarfism. It presents with an exceptionally short stature, microcephaly, and unique facial attributes. The features that distinguish it from other primordial dwarfism types, which might require medical attention, encompass irregular dental structures, a fragile bone skeletal dysplasia leading to hip deformities or scoliosis, tendencies towards insulin resistance or diabetes, chronic kidney ailments, heart defects, and a broad spectrum of vascular diseases. This wide-ranging vascular ailment category encompasses neurovascular conditions like moyamoya vasculopathy and intracranial aneurysms (potentially causing strokes), early-onset coronary artery diseases (potentially resulting in premature heart attacks), and kidney-related vascular issues. The frequent occurrence of hypertension is complicated due to multiple potential origins tied to the array of associated conditions [22][21].
Observationally, from birth onwards, 25 out of 47 individuals were identified with intracranial aneurysms (30 of them had multiple aneurysms), 22 showed signs of moyamoya vasculopathy, 17 exhibited both conditions, and 17 had neither. Roughly 50% of those diagnosed with either moyamoya vasculopathy or aneurysms eventually experienced a stroke, be it ischemic or hemorrhagic. The risk of an aneurysm remained fairly consistent throughout childhood, while the risk associated with moyamoya vasculopathy was accentuated before the age of five [23][22].
Type IV Ehlers–Danlos Syndrome (Vascular Subtype): This autosomal dominant condition, affecting between 1 in 50,000 to 200,000 individuals, is a connective tissue disorder. It is chiefly marked by pronounced vascular fragility, heightening the risk of a hemorrhage and premature death [24][23]. The underlying cause of vascular EDS is the presence of mutations or variants in the Col3a1 gene. This gene is responsible for producing a protein that plays a crucial role in reinforcing connective tissue throughout the body [25][24].
Microcephalic/Majewski’s Osteodysplastic Primordial Dwarfism, Type II (MOPD2): This exceedingly rare autosomal recessive disorder is characterized by a notably short stature, alongside skeletal abnormalities, especially a significantly small head size in proportion to the body [26][25]. Given the scant occurrence of MOPD2, establishing a precise prevalence of IA in affected individuals remains challenging. However, current evidence indicates that as many as half of those with MOPD2 might develop an IA [27][26]. The genetic basis for MOPD2 is traced back to mutations in the Pericentrin 1 (PCNT) gene. This gene encodes for a protein that is central to chromosomal segregation. Additionally, a loss-of-function in PCNT disrupts cilia formation in epithelial cells and impacts PDK2 positioning at the basal bodies [28][27]. The intricate interaction between PCNT and PDK2 and the potential implications of PCNT insufficiency in an IA becomes even more evident when considering the presence of rare PCNT variants in multiple IA-affected families. Some family members also presented with kidney cysts [29][28].
However, in these families, it is crucial to understand that other genetic variants, apart from PCNT, may exist. These additional variants might play significant roles in disease manifestation and progression.
Loeys–Dietz Syndrome (LDS): LDS shares similarities with type IV Ehlers–Danlos, being an autosomal dominant connective tissue disease. Caused by mutations in the genes of the Tgf-β pathway (mainly TGFBR1, TGFBR2, SMAD3, and TGFB2), patients exhibit severe vascular anomalies. Arterial aneurysm dissections and bleeding events, which often appear early in life, are frequent complications. A significant portion, roughly one-third, of LDS-related deaths, result from cerebrovascular hemorrhages. The incidence of an IA among those with LDS ranges from 10% to 28% [30][29].
Marfan Syndrome is among the prevalent hereditary disorders impacting connective tissue. It is an autosomal dominant condition, manifesting in approximately 1 out of every 3000 to 5000 individuals. The underlying genetic flaw is in the FBN1 gene on chromosome 15, which is responsible for producing fibrillin, a vital connective tissue protein. The heightened risk associated with the aorta in males has been recognized for a while, yet its underlying reason remains unclear. In this context, the influence of gender was evident across all molecular categories. Patients with PTC displayed a trend towards earlier surgeries, and the lifetime aortic risk for males was double compared to females who possessed in-frame pathogenic variations [31][30]. Though some studies suggest a link between Marfan’s syndrome and IAs, autopsies and family analyses with multiple members having both an IA and Marfan’s syndrome do not consistently support this association [32,33][31][32].
Neurofibromatosis type 1: NF1 is a condition marked by tumors in the skin and nervous system. The two primary forms, types 1 and 2, are both inherited in an autosomal dominant manner. Type 1, often referred to as von Recklinghausen disease, is typified by features like neurofibromas, cafe-au-lait spots, freckling, and optic gliomas. In contrast, type 2 is distinguished by the presence of bilateral vestibular schwannomas and meningiomas [34][33].
In a 2005 research study by Schievink et al. [35][34], 39 patients with neurofibromatosis type 1 were examined, with an average age of 30.4 years, ranging from 3 to 64 years. Incidental intracranial aneurysms were found in 2 (5%) out of the 39 patients through MRI scans. When focusing only on the 22 patients who had an MRI, the detection rate rose to 9%. This rate was notably higher (p < 0.005) than the 0% detection rate in the control group of 526 individuals, therefore concluding the existence of a correlation between NF1 as a genetic risk factor for IA development.
CDKN2BAS: The 9p21.3 locus first gained attention during a 2007 genome-wide association study (GWAS) related to cardiovascular disease. In this study, it was highlighted that the specific genetic variations associated with cardiovascular diseases also had a connection with intracranial aneurysms (IAs). Interestingly, the 9p21.3 region itself does not house any known protein-coding genes. However, in its vicinity—just a few kilobases away—are protein-coding genes, including CDKN2A and CDKN2B [36][35]. It is worth mentioning that the 9p21.3 gene cluster (CDKN2A-CDKN2B) has emerged as a frequently identified region in GWASs for several prevalent conditions, such as heart diseases, type 2 diabetes, brain tumors (gliomas), and skin cancers (basal cell carcinomas) [37][36].
One significant discovery came from a study that pinpointed the CDKN2BAS SNP (rs6475606) as a contributing factor for IA vulnerability, particularly in the US population. This was a monumental finding, as understanding genetic predispositions can be the key to predicting and potentially preventing the onset of conditions like IAs [38][37].
Subsequent GWAS research involving both Asian and Caucasian populations has further underscored this genetic link. Numerous single-nucleotide polymorphisms (SNPs) of CDKN2BAS have been identified as being associated with intracranial aneurysms. This consistent finding across diverse populations suggests a robust genetic connection and underscores the importance of this genetic region in understanding, and potentially addressing, IAs [39][38].
Other Syndromes: Several conditions, such as multiple endocrine neoplasia type I [40][39], pseudoxanthoma elasticum [41][40], and hereditary hemorrhagic telangiectasia (HHT) [42][41], are frequently highlighted in relation to intracranial berry aneurysm. While there are individual patient case studies documenting the existence of intracranial aneurysms in these conditions, the evidence is not as comprehensive as it is for other syndromes that were previously discussed [43][42].
2.2. Genetic Implications
A predominant strategy for discerning genetic risk factors related to intracranial aneurysms (IAs) entails genotyping polymorphisms within sporadic cases and controls. The genetic markers frequently employed in these endeavors are termed single nucleotide polymorphisms (SNPs). Genetic association investigations can be segmented into two main categories: Candidate Gene Association Studies (CGASs) and genome-wide association studies (GWASs). A CGAS involves the examination of select common polymorphisms in genes, specifically chosen based on existing biological evidence suggesting their relevance to IA development. Additionally, candidate genes for IAs have been sourced from genetic studies focusing on connective tissue disorders like vascular Ehlers–Danlos syndrome; known genetic diseases where an IA is a manifestation, such as polycystic kidney disease; and from gene expression studies [44][43].
A plethora of recognized mendelian disorders is known to elevate the risk of IA development. Over recent decades, there has been a recalibration in the understanding of genetic causation. Previously, the paradigm centered on single gene mutations that are directly correlated with distinct clinical phenotypes. However, the contemporary perspective emphasizes the intricate interactions between singular gene mendelian phenotypes and allelic variants located elsewhere in the genome, termed transacting regulatory elements. Consequently, the understanding of genetic inheritance in diseases has evolved from a binary approach to a more nuanced, spectrum-oriented view [45][44].
The proclivity towards IAs, stemming from these genetic anomalies, likely emanates from structural deficiencies in the brain’s vasculature due to the malfunctioning of specific genes. In illuminating this, a recent meta-analysis concerning the ACE insertion/deletion (I/D) polymorphism (rs4646994) demonstrated a linkage between the I allele and increased IA susceptibility. Specifically, individuals possessing ACE I/I and I/D genetic configurations displayed a heightened IA risk. The exact mechanisms by which the ACE I allele influences the development of an IA remain nebulous [46][45].
Enriching the discourse, Yasuno and colleagues amalgamated more datasets, thereby analyzing a larger sample consisting of 5891 cases and 14,181 controls. Their findings revealed a total of five IA-specific loci. Among these, the SOX17 (8q11.23; rs9298506) and CDKN2BAS1 (9p21; rs1333040) loci were previously identified, and their association with IAs was fortified in this research. Although the SNP rs10958409 (8q11.23; rs10958409) did not manifest an association, the study unveiled three novel IA associations with CNNM2 (10q24.32; rs12413409), STARD13 (13q13.1; rs9315204), and RBBP8 (18q11.2; rs11661542). It is pivotal to acknowledge that certain SNPs, despite achieving genome-wide significance like rs700651 and rs11661542, did not exhibit replicability in subsequent studies and thus were not deemed significant in the encompassing meta-analyses [47][46] (Figure 1 and Table 1).
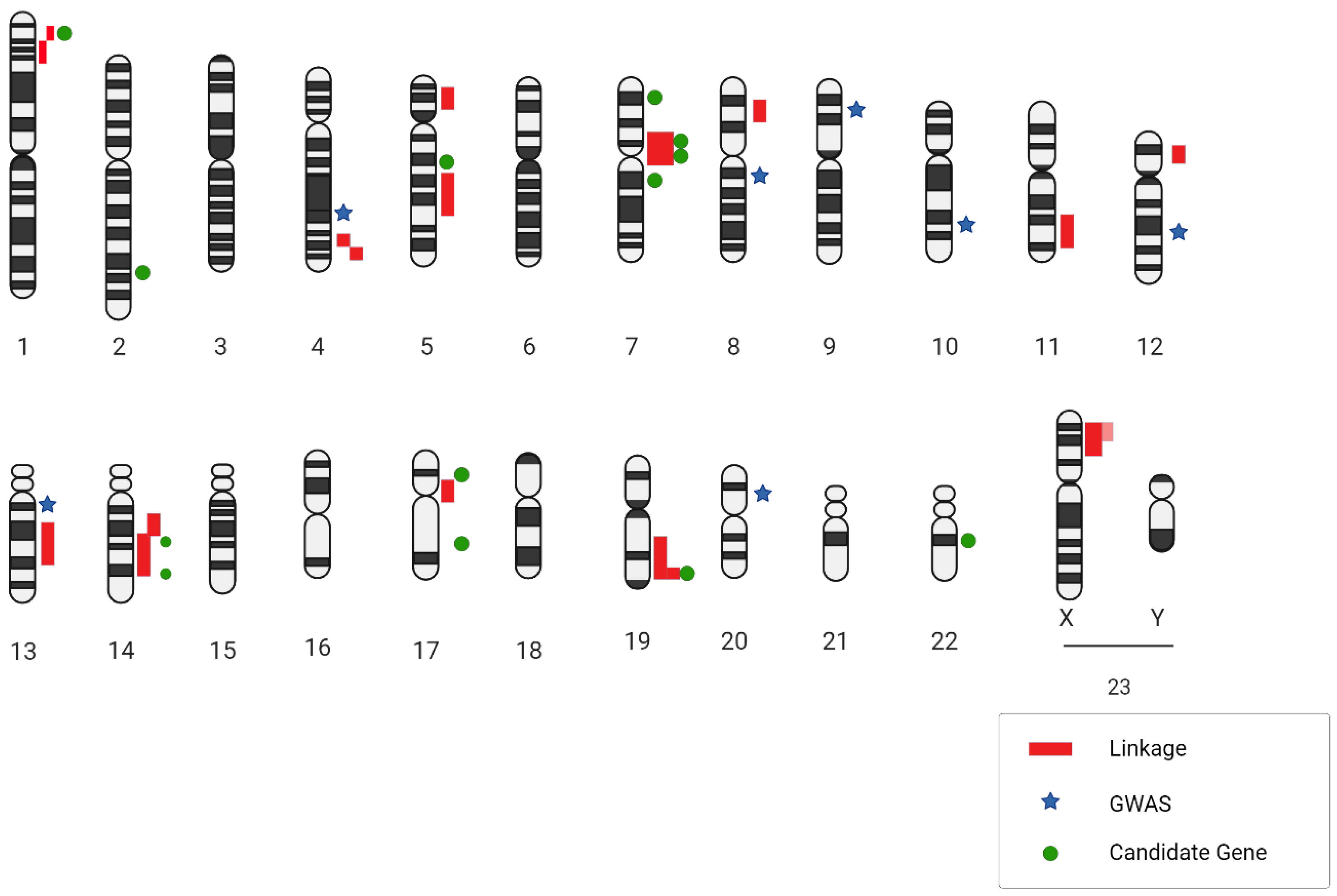
Figure 1. A genetic blueprint highlighting locations related to intracranial aneurysms is presented. Next to the chromosome diagrams, dark red lines point out regions identified through DNA linkage research. Light red lines suggest regions supported by family links to two specific spots. Blue star symbols mark the places where SNPs were identified in wide-ranging genetic studies, while green circular markers show where SNPs were pinpointed in targeted gene association studies.
Table 1.
Linkage analyses of DNA related to intracranial aneurysms.
| Chromosomal Region | Study Design | LOD Score | Genetic Marker | Phenotype IDs and OMIM Locus |
|---|---|---|---|---|
| 1p36.21-p36.13 | Non-parametric | 3.18 | D1S2826-D1S234 | 609122; ANIB3 |
| 1p34.21-p36.13 | Family, AD | 4.2 | 609122; ANIB3 | |
| 4q32.2 | Non-parametric | 2.5 | Rs1458149 | |
| 4q32.3 | Parametric | 2.6 | ||
| 5p15.2-p14.3 | Family, AD | 3.57 | D5S1954 | 610213; ANIB4 |
| 5q22-q31 | Affected sib pair | 2.24 | D5S471-D5S2010 | |
| 7q11 | AR | 2.34 | D7S2421 | 105800; ANIB1 |
| 7q11 | Affected sib pair | 3.22 | D7S2415-D7S657 | 105800; ANIB1 |
| 8p22 | Family, AD | 3.61 | ||
| DXS6807-DXS1224 | ||||
| 300870; ANIB5 | ||||
AD = autosomal dominant; AR = autosomal recessive.
There exist certain genetic syndromes that are transmitted through an autosomal recessive manner or stem from spontaneous genetic alterations. These are known to be associated with intracranial aneurysms, albeit at a lesser frequency compared to more prevalent syndromes. An exemplar of such conditions is pseudoxanthoma elasticum. This disorder is linked to mutations found within the ABCC6 gene situated on chromosome 16p13 [48][47]. One prevailing theory proposes that IAs in individuals with this syndrome originate due to irregularities in the elastic lamina of the intracranial blood vessels, with the intracranial internal carotid artery being a primary site [41][40]. Whole exome sequencing (WES) has spotlighted the potential significance of the PCNT gene in relation to cerebrovascular diseases. The protein encoded by this gene plays a pivotal role in orchestrating microtubule nucleation and ensuring the orderly progression of the cell cycle. Importantly, mutations where both copies of the gene are dysfunctional lead to a condition known as microcephalic osteodysplastic primordial dwarfism type II. Astonishingly, in over half of these cases, it is surmised that individuals bearing specific mutations in the PCNT gene are susceptible to IAs. Intriguingly, these genetic variations might also amplify the risk of a single individual manifesting multiple IAs [27][26].
While these genetic anomalies offer invaluable insights into the genesis of aneurysms, comprehending the specific risks tied to aneurysm rupture in these distinct syndromes necessitates further exploration. Beyond merely identifying the genetic markers related to the onset or presence of IAs, there exists a trove of data ripe for analysis to pinpoint genetic locations that might dictate the propensity for aneurysmal rupture and the subsequent clinical aftermath of a subarachnoid hemorrhage (SAH). The post-rupture genetic landscape of the disease becomes murkier, as there could be other genetic factors influencing various aspects of SAH care. One such aspect is vasospasm—a contraction of blood vessels—which is widely acknowledged as a key contributor to grievous outcomes due to delayed cerebral ischemia (DCI) [49][48]. Presently, there is a noticeable scarcity of comprehensive studies elucidating outcomes post aneurysm rupture. The limited studies available in the scientific literature often lack the rigorous sample sizes required for drawing incontrovertible conclusions. Most of these outcome-centric studies place their emphasis on the ever-fluctuating gene expression profiles. This dynamic nature contrasts with static genetic anomalies, which could potentially offer a constant measure of risk, hence posing challenges in consistently assessing the genetic basis of the outcomes [50][49].
2.3. Genome-Wide Association Studies
Genome-wide linkage analyses are sophisticated methods utilized to pinpoint the specific chromosomal location, referred to as loci, that houses the gene that is responsible for certain diseases. To accomplish this, these analyses probe specific genetic markers, such as single nucleotide polymorphism (SNP) and Variable Number Tandem Repeat (VNTR) [51][50]. Thanks to the versatility of the linkage analysis method, researchers have managed to unveil genes that drive the onset of complex disorders. These range from metabolic conditions like diabetes and obesity to cardiovascular issues such as hypertension. The breakthroughs provided by these analyses have deepened the understanding of the genetic underpinnings of such multifaceted ailments [52,53][51][52]. Over time, numerous studies have leveraged linkage analysis to unearth insights about IAs. The allure of non-parametric methodologies is particularly potent when it comes to IAs. This is primarily because the penetrance—referring to the likelihood that an individual with a mutation will display symptoms—may not be absolute. In simpler terms, not every individual harboring the mutation will be afflicted. Summarily, non-parametric linkage studies have unveiled several loci that may play roles in the formation and potential rupture of IAs. However, only a select few, namely 1p34.3-p36.13, 7q11, 19q13.3, and Xp22, have seen consistent results across various populations [54,55,56][53][54][55].
Current research furnishes the most compelling evidence concerning regions on chromosomal arms 7q and 19q. Numerous independent studies have reported linkage hits on these arms, signaling their potential significance in IAs. Alg et al., in a comprehensive meta-analysis involving a staggering 116,000 participants, spotlighted 19 SNPs that exhibited a significant correlation with IAs under at least one genetic model. GWASs emerged as particularly illuminating, uncovering 12 robustly associated SNPs. Some of the most telling associations are related to chromosomes 9, 8, and 4. Meanwhile, the CGAS method identified eight significant SNPs, with SERPINA3 and two collagen-related variants showcasing the strongest links. Notably, 9 out of the 19 SNPs exhibited significant statistical variance, mandating random-effects and sensitivity evaluations for those reported in over two publications [57][56].
A recent genome-wide association study, centered on both familial and sporadic IAs in a Caucasian population, identified six SNPs in the 9p21.3 region as being significantly associated with IA. Of these, one (rs6475606) achieved a stellar level of statistical significance for its association with both types of IAs. Additionally, the study reaffirmed the association of the rs1333040 SNP with IAs, bolstering the belief in its relevance [58][57].
The endothelin receptor type A (often abbreviated as EDNRA) gene encodes a receptor that is activated by endothelins, a group of proteins instrumental in regulating both the constriction and dilation of blood vessels, particularly following any hemodynamic disturbances. Specifically, Endothelin-1, which is the primary variant present in vascular smooth muscle cells, is responsible for triggering EDNRA. This endothelin signaling pathway becomes particularly active at sites of vascular injuries, leading to heightened cell proliferation—a critical step in the body’s reparative process [59][58]. There is a potential that a diminished activity, or downregulation, of EDNRA signaling might be a precursor to compromised vascular repair mechanisms. When this repair process does not function optimally, it could make the vascular system more susceptible to the formation of aneurysms following injuries or disturbances. Adding credence to this theory is the functional analysis of the EDNRA gene variants. Specifically, the rs6841581 risk allele has been shown to have reduced transcriptional activity. This essentially means that the gene’s ability to produce its associated protein might be diminished, which could potentially contribute to the aforementioned vulnerabilities in the vascular repair system [60][59].
Continuing research using genome-wide association studies recently highlighted another intriguing discovery. The SNP denoted as rs6841581A.G, located on chromosome 4q31.23, which codes for the EDNRA gene, has caught researchers’ attention. This particular SNP displayed a significant association with intracranial aneurysms. What is particularly noteworthy is that this association was not restricted to just one ethnic group or population; it emerged as a consistent pattern across Dutch, Finnish, and Japanese populations. This widespread correlation accentuates the potential global relevance and importance of the EDNRA gene in understanding and potentially addressing IAs [61][60] (Table 2).
Table 2.
Key research findings on intracranial aneurysm genetics: loci, candidate genes, and associated functions.
| Type of Genetic Analysis | Loci | Candidate Genes | Functions | |||||
|---|---|---|---|---|---|---|---|---|
| GWAS | 13q13.1 | STARD 13 | Movement of endothelial cells | |||||
| GWAS | 18q11.2 | RBBP8 | Cell cycle | |||||
| GWAS | 2q | PLCL1, BOLL | Similarity to phospholipase C, positioned after VEGFR2 in the signaling pathway | |||||
| GWAS | 4q31.23 | EDNRA | Vasoconstriction | |||||
| GWAS | 8q21.3 | SOX17 | Endothelial sprouting | |||||
| GWAS | 9p21.3 | CDKN2A/B | Smooth muscle proliferation | |||||
| GWAS | 10q24.32 | CNNM2 | Epithelial absorption of Mg2+ | |||||
| GWL | 1p34.3-p36.13Xp22 | PERLECAN | Promote endothelial cell growth and renewal, maintain the endothelial barrier function, and inhibit smooth muscle cell proliferation | |||||
| D8S552 | 614252; ANIB11 | |||||||
| GWL | 7q11, 14q22, 5q22 | ELASTIN | Elasticity of the parietal vessel | 11q24-q25 | Family | 4.3 | rs618176-rs1940033 | 612161; ANIB7 |
| 12p12.3 | ||||||||
| GWL | 11q24, 14q23 | Parametric | 3.1 | |||||
| 13q14.12-q21.1 | Family | 4.56 | rs7983420-rs17054625 | |||||
| 14q23-q31 | Family | 3.0 | rs235991-rs2373098 | 612162; ANIB8 | ||||
| 14q22 | Affected sib pair | 2.31 | D14S258-D14S74 | |||||
| 17cen | Non-parametric | 3.0 | D17S921-D17S1800 | |||||
| 19q13 | Non-parametric | 2.15 | D19S198-D19S596 | 608542; ANIB2 | ||||
| 19q13 | Affected sib pair with covariates | 5.70 | D19S178-D19S545 | 608542; ANIB2 | ||||
| 19q13.3 | ||||||||
| GWL | 19q13, Xp22 | Affected only, parametric | 4.10 | D19S198-D19S902 | 608542; ANIB2 | |||
| Xp22 | Non-parametric | 2.16 | DXS987-DXS7593 | 300870; ANIB5 | ||||
| Xp22 | Affected sib pair | 2.08 | DXS987 | 300870; ANIB5 | ||||
| Xp22.32-p22.2 | Non-parametric | 4.54 | ||||||
Abbreviations: GWL = genome wide linkage; GWAS = genome wide association studies.
3. Molecular and Physiopathological Mechanisms Implicated in IA Formation and Progression
Human IA samples serve as a treasure trove for researchers, offering them a unique glimpse into the intricate molecular mechanisms underpinning an IA’s onset and rupture. At the heart of IA pathology is the degradation of the internal elastic lamina (IEL), which sits as a protective barrier separating the intima from the media layers of blood vessels. Alongside this, various other signs, like an uneven vessel inner surface, thickening due to myointimal growth, the chaotic nature of the muscular media, reduced cell presence, and heightened inflammatory cell infiltration, paint a complex picture of an IA’s internal environment [62][61].
As a critical component in arteries, the IEL comprises elastic fibers that grant flexibility to these vessels. In a healthy state, the IEL presents as consistent and intact in intracranial arteries. However, when confronted with an IA, the IEL tends to become fragmented or torn, or may even vanish completely, which is especially evident at the aneurysm’s base. This degradation is a significant red flag pointing towards an IA’s pathology [63][62].
One of the more striking aspects of IAs, whether from human patients or those mimicked in animal models, is the formation of pronounced outward bulges and deep inward crevices in the intimal lining. These anomalies become evident through the intricate imaging of transmission electron microscopy [64][63].
The degradation of the IEL acts as a trigger. It allows for the migration of smooth muscle cells from the media to the intima layer. Here, they multiply, leading to myointimal hyperplasia, resulting in a pronounced thickening of the intima layer, which is a recurring characteristic in IA samples [65][64].
IAs mark a shift in the medial layer’s cellular configuration. Here, smooth muscle cells transition or “switch” from their primary contractile state to a more synthetic one. This new state is marked by heightened inflammatory and remodeling tendencies. In the context of an IA, there is a heightened influx of inflammatory cells. Both animal models and human samples consistently reveal the presence of cells like macrophages, T cells, B cells, and neutrophils. Significantly, macrophages, apart from their standard functions, release matrix-degrading enzymes such as MMP2 and MMP9. These enzymes, alongside various cytokines, serve to attract a greater volume of inflammatory cells to the site [64][63].
Microarray-based mRNA profiling stands out as a holistic tool, allowing for the clear demarcation of molecular markers in both healthy and diseased states. While arteries have their distinct mRNA expression profiles, the same type of artery from varied individuals tends to have more similarities than different arteries within one individual. This highlights the necessity for meticulous control tissue selection when conducting expression studies [66][65].
To truly discern the molecular markers specific to an intracranial artery, researchers utilized RNA samples from both IA-affected and unaffected arteries, applying two microarray platforms. Intriguingly, almost half of the genes on these platforms were expressed in these arteries. Further, DNA linkage studies spotlighted around 800 diverse genes present in intracranial arteries. Utilizing advanced bioinformatics tools like GO and KEGG, researchers found a significant clustering around biological pathways, including the likes of Notch and MAPK signaling channels [67][66].
Genome-wide studies focusing on both mRNA and miRNA expressions offer a comprehensive, unbiased pathway to dive into an IA’s pathophysiology. Tools like GO and KEGG not only facilitate a more structured understanding of gene expression, but also throw light on the intricate web of interconnected pathways, providing a holistic view of the underlying pathology defining an IA.
References
- Brown, R. Unruptured Intracranial Aneurysms. Semin. Neurol. 2010, 30, 537–544.
- Rinkel, G.J.E.; Djibuti, M.; Algra, A.; Van Gijn, J. Prevalence and Risk of Rupture of Intracranial Aneurysms: A Systematic Review. Stroke 1998, 29, 251–256.
- Levy, M.L.; Levy, D.M.; Manna, B. Pediatric Cerebral Aneurysm. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. Available online: http://www.ncbi.nlm.nih.gov/books/NBK537085/ (accessed on 10 August 2023).
- Menghini, V.V.; Brown, R.D.; Sicks, J.D.; O’Fallon, W.M.; Wiebers, D.O. Incidence and prevalence of intracranial aneurysms and hemorrhage in Olmsted County, Minnesota, 1965 to 1995. Neurology 1998, 51, 405–411.
- Ujiie, H.; Sato, K.; Onda, H.; Oikawa, A.; Kagawa, M.; Takakura, K.; Kobayashi, N. Clinical analysis of incidentally discovered unruptured aneurysms. Stroke 1993, 24, 1850–1856.
- The International Study of Unruptured Intracranial Aneurysms Investigators. Unruptured Intracranial Aneurysms—Risk of Rupture and Risks of Surgical Intervention. N. Engl. J. Med. 1998, 339, 1725–1733.
- Schackert, H.K.; Schackert, G.; Krex, D. Genesis of Cerebral Aneurysms—An Update. Acta Neurochir. 2001, 143, 429–449.
- Nakagawa, T.; Hashi, K. The incidence and treatment of asymptomatic, unruptured cerebral aneurysms. J. Neurosurg. 1994, 80, 217–223.
- De Rooij, N.K.; Linn, F.H.H.; Van Der Plas, J.A.; Algra, A.; Rinkel, G.J.E. Incidence of subarachnoid haemorrhage: A systematic review with emphasis on region, age, gender and time trends. J. Neurol. Neurosurg. Psychiatry 2007, 78, 1365–1372.
- Johnston, S.C.; Selvin, S.; Gress, D.R. The burden, trends, and demographics of mortality from subarachnoid hemorrhage. Neurology 1998, 50, 1413–1418.
- Grobelny, T.J. Brain Aneurysms: Epidemiology, Treatment Options, and Milestones of Endovascular Treatment Evolution. Dis. Mon. 2011, 57, 647–655.
- Kubota, M.; Yamaura, A.; Ono, J. Prevalence of risk factors for aneurysmal subarachnoid haemorrhage: Results of a Japanese multicentre case control study for stroke. Br. J. Neurosurg. 2001, 15, 474–478.
- Clarke, M. Systematic review of reviews of risk factors for intracranial aneurysms. Neuroradiology 2008, 50, 653–664.
- Kernan, W.N.; Viscoli, C.M.; Brass, L.M.; Broderick, J.P.; Brott, T.; Feldmann, E.; Morgenstern, L.B.; Wilterdink, J.L.; Horwitz, R.I. Phenylpropanolamine and the Risk of Hemorrhagic Stroke. N. Engl. J. Med. 2000, 343, 1826–1832.
- Greving, J.P.; Wermer, M.J.H.; Brown, R.D.; Morita, A.; Juvela, S.; Yonekura, M.; Ishibashi, T.; Torner, J.C.; Nakayama, T.; Rinkel, G.J.E.; et al. Development of the PHASES score for prediction of risk of rupture of intracranial aneurysms: A pooled analysis of six prospective cohort studies. Lancet Neurol. 2014, 13, 59–66.
- Raaymakers, T.W.M. Aneurysms in relatives of patients with subarachnoid hemorrhage: Frequency and risk factors. Neurology 1999, 53, 982.
- Cagnazzo, F.; Gambacciani, C.; Morganti, R.; Perrini, P. Intracranial aneurysms in patients with autosomal dominant polycystic kidney disease: Prevalence, risk of rupture, and management. A systematic review. Acta Neurochir. 2017, 159, 811–821.
- Pirson, Y. Extrarenal Manifestations of Autosomal Dominant Polycystic Kidney Disease. Adv. Chronic Kidney Dis. 2010, 17, 173–180.
- Vlak, M.H.; Algra, A.; Brandenburg, R.; Rinkel, G.J. Prevalence of unruptured intracranial aneurysms, with emphasis on sex, age, comorbidity, country, and time period: A systematic review and meta-analysis. Lancet Neurol. 2011, 10, 626–636.
- Harburger, D.S.; Calderwood, D.A. Integrin signalling at a glance. J. Cell Sci. 2009, 122, 159–163.
- Duker, A.; Jackson, A.; Bober, M.B. Microcephalic Osteodysplastic Primordial Dwarfism Type II. In GeneReviews®; Adam, M.P., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. Available online: http://www.ncbi.nlm.nih.gov/books/NBK575926/ (accessed on 10 August 2023).
- Duker, A.L.; Kinderman, D.; Jordan, C.; Niiler, T.; Baker-Smith, C.M.; Thompson, L.; Parry, D.A.; Carroll, R.S.; Bober, M.B. Microcephalic osteodysplastic primordial dwarfism type II is associated with global vascular disease. Orphanet J. Rare Dis. 2021, 16, 231.
- De Paepe, A.; Malfait, F. The Ehlers-Danlos syndrome, a disorder with many faces. Clin. Genet. 2012, 82, 1–11.
- Superti-Furga, A.; Gugler, E.; Gitzelmann, R.; Steinmann, B. Ehlers-Danlos syndrome type IV: A multi-exon deletion in one of the two COL3A1 alleles affecting structure, stability, and processing of type III procollagen. J. Biol. Chem. 1988, 263, 6226–6232.
- Bober, M.B.; Khan, N.; Kaplan, J.; Lewis, K.; Feinstein, J.A.; Scott, C.I.; Steinberg, G.K. Majewski Osteodysplastic Primordial Dwarfism Type II (MOPD II): Expanding the vascular phenotype. Am. J. Med. Genet. A 2010, 152A, 960–965.
- Li, F.-F.; Wang, X.-D.; Zhu, M.-W.; Lou, Z.-H.; Zhang, Q.; Zhu, C.-Y.; Feng, H.-L.; Lin, Z.-G.; Liu, S.-L. Identification of two novel critical mutations in PCNT gene resulting in microcephalic osteodysplastic primordial dwarfism type II associated with multiple intracranial aneurysms. Metab. Brain Dis. 2015, 30, 1387–1394.
- Jurczyk, A.; Gromley, A.; Redick, S.; Agustin, J.S.; Witman, G.; Pazour, G.J.; Peters, D.J.M.; Doxsey, S. Pericentrin forms a complex with intraflagellar transport proteins and polycystin-2 and is required for primary cilia assembly. J. Cell Biol. 2004, 166, 637–643.
- Lorenzo-Betancor, O.; Blackburn, P.R.; Edwards, E.; Vázquez-do-Campo, R.; Klee, E.W.; Labbé, C.; Hodges, K.; Glover, P.; Sigafoos, A.N.; Soto, A.I.; et al. PCNT point mutations and familial intracranial aneurysms. Neurology 2018, 91, e2170–e2181.
- Kim, S.T.; Brinjikji, W.; Kallmes, D.F. Prevalence of Intracranial Aneurysms in Patients with Connective Tissue Diseases: A Retrospective Study. Am. J. Neuroradiol. 2016, 37, 1422–1426.
- Arnaud, P.; Milleron, O.; Hanna, N.; Ropers, J.; Ould Ouali, N.; Affoune, A.; Langeois, M.; Eliahou, L.; Arnoult, F.; Renard, P.; et al. Clinical relevance of genotype–phenotype correlations beyond vascular events in a cohort study of 1500 Marfan syndrome patients with FBN1 pathogenic variants. Genet. Med. 2021, 23, 1296–1304.
- Conway, J.E.; Hutchins, G.M.; Tamargo, R.J. Marfan Syndrome Is Not Associated With Intracranial Aneurysms. Stroke 1999, 30, 1632–1636.
- Van Den Berg, J.S.P.; Limburg, M.; Hennekam, R.C.M. Is Marfan Syndrome Associated With Symptomatic Intracranial Aneurysms? Stroke 1996, 27, 10–12.
- Le, C.; Bedocs, P.M. Neurofibromatosis. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. Available online: http://www.ncbi.nlm.nih.gov/books/NBK459329/ (accessed on 10 August 2023).
- Schievink, W.I.; Riedinger, M.; Maya, M.M. Frequency of incidental intracranial aneurysms in neurofibromatosis type 1. Am. J. Med. Genet. Part A 2005, 134A, 45–48.
- Broadbent, H.M.; Peden, J.F.; Lorkowski, S.; Goel, A.; Ongen, H.; Green, F.; Clarke, R.; Collins, R.; Franzosi, M.G.; Tognoni, G.; et al. Susceptibility to coronary artery disease and diabetes is encoded by distinct, tightly linked SNPs in the ANRIL locus on chromosome 9p. Hum. Mol. Genet. 2008, 17, 806–814.
- Zhou, S.; Dion, P.A.; Rouleau, G.A. Genetics of Intracranial Aneurysms. Stroke 2018, 49, 780–787.
- Salvi, E.; Kutalik, Z.; Glorioso, N.; Benaglio, P.; Frau, F.; Kuznetsova, T.; Arima, H.; Hoggart, C.; Tichet, J.; Nikitin, Y.P.; et al. Genomewide Association Study Using a High-Density Single Nucleotide Polymorphism Array and Case-Control Design Identifies a Novel Essential Hypertension Susceptibility Locus in the Promoter Region of Endothelial NO Synthase. Hypertension 2012, 59, 248–255.
- Yu, T.; Jiang, H.; Fan, Y.; Xu, Y.; Wang, N. The association of CDKN2BAS gene polymorphisms and intracranial aneurysm: A meta-analysis. Medicine 2020, 99, e23209.
- Adachi, K.; Kudo, M.; Chen, M.N.; Nakazawa, S.; Wakabayashi, I. Cerebral Aneurysm Associated with Multiple Endocrine Neoplasia, Type 1—Case Report. Neurol. Med. Chir. 1993, 33, 309–311.
- Bock, A.; Schwegler, G. Intracerebral haemorrhage as first manifestation of Pseudoxanthoma elasticum. Clin. Neurol. Neurosurg. 2008, 110, 262–264.
- Willemse, R.B.; Mager, J.J.; Westermann, C.J.J.; Overtoom, T.T.C.; Mauser, H.; Wolbers, J.G. Bleeding risk of cerebral vascular malformations in hereditary hemorrhagic telangiectasia. J. Neurosurg. 2000, 92, 779–784.
- Hitchcock, E.; Gibson, W.T. A Review of the Genetics of Intracranial Berry Aneurysms and Implications for Genetic Counseling. J. Genet. Couns. 2017, 26, 21–31.
- Tromp, G.; Weinsheimer, S.; Ronkainen, A.; Kuivaniemi, H. Molecular basis and genetic predisposition to intracranial aneurysm. Ann. Med. 2014, 46, 597–606.
- Nadeau, J.H. Modifier genes in mice and humans. Nat. Rev. Genet. 2001, 2, 165–174.
- You, C.; Chen, Z.; Ma, J.; Cen, Y.; Liu, Y. The angiotensin converting enzyme insertion/deletion polymorphism and intracranial aneurysm: A meta-analysis of case-control studies. Neurol. India 2013, 61, 293.
- Yasuno, K.; Bilguvar, K.; Bijlenga, P.; Low, S.-K.; Krischek, B.; Auburger, G.; Simon, M.; Krex, D.; Arlier, Z.; Nayak, N.; et al. Genome-wide association study of intracranial aneurysm identifies three new risk loci. Nat. Genet. 2010, 42, 420–425.
- Moitra, K.; Garcia, S.; Jaldin, M.; Etoundi, C.; Cooper, D.; Roland, A.; Dixon, P.; Reyes, S.; Turan, S.; Terry, S.; et al. ABCC6 and Pseudoxanthoma Elasticum: The Face of a Rare Disease from Genetics to Advocacy. Int. J. Mol. Sci. 2017, 18, 1488.
- Samuel, N.; Radovanovic, I. Genetic basis of intracranial aneurysm formation and rupture: Clinical implications in the postgenomic era. Neurosurg. Focus 2019, 47, E10.
- Vergouwen, M.D.I.; Frijns, C.J.M.; Roos, Y.B.W.E.M.; Rinkel, G.J.E.; Baas, F.; Vermeulen, M. Plasminogen Activator Inhibitor-1 4G Allele in the 4G/5G Promoter Polymorphism Increases the Occurrence of Cerebral Ischemia After Aneurysmal Subarachnoid Hemorrhage. Stroke 2004, 35, 1280–1283.
- Xiang, C.; Liu, S.; Fan, Y.; Wang, X.; Jia, Y.; Li, L.; Cong, S.; Han, F. Single nucleotide polymorphisms, variable number tandem repeats and allele influence on serotonergic enzyme modulators for aggressive and suicidal behaviors: A review. Pharmacol. Biochem. Behav. 2019, 180, 74–82.
- Concannon, P.; Erlich, H.A.; Julier, C.; Morahan, G.; Nerup, J.; Pociot, F.; Todd, J.A.; Rich, S.S. Type 1 Diabetes: Evidence for Susceptibility Loci from Four Genome-Wide Linkage Scans in 1,435 Multiplex Families. Diabetes 2005, 54, 2995–3001.
- Wilson, F.H.; Disse-Nicodème, S.; Choate, K.A.; Ishikawa, K.; Nelson-Williams, C.; Desitter, I.; Gunel, M.; Milford, D.V.; Lipkin, G.W.; Achard, J.-M.; et al. Human Hypertension Caused by Mutations in WNK Kinases. Science 2001, 293, 1107–1112.
- Nahed, B.V.; Seker, A.; Guclu, B.; Ozturk, A.K.; Finberg, K.; Hawkins, A.A.; DiLuna, M.L.; State, M.; Lifton, R.P.; Gunel, M. Mapping a Mendelian Form of Intracranial Aneurysm to 1p34.3-p36.13. Am. J. Hum. Genet. 2005, 76, 172–179.
- Yamada, S.; Utsunomiya, M.; Inoue, K.; Nozaki, K.; Inoue, S.; Takenaka, K.; Hashimoto, N.; Koizumi, A. Genome-Wide Scan for Japanese Familial Intracranial Aneurysms: Linkage to Several Chromosomal Regions. Circulation 2004, 110, 3727–3733.
- Onda, H.; Kasuya, H.; Yoneyama, T.; Takakura, K.; Hori, T.; Takeda, J.; Nakajima, T.; Inoue, I. Genomewide-Linkage and Haplotype-Association Studies Map Intracranial Aneurysm to Chromosome 7q11. Am. J. Hum. Genet. 2001, 69, 804–819.
- Alg, V.S.; Sofat, R.; Houlden, H.; Werring, D.J. Genetic risk factors for intracranial aneurysms: A meta-analysis in more than 116,000 individuals. Neurology 2013, 80, 2154–2165.
- Foroud, T.; Koller, D.L.; Lai, D.; Sauerbeck, L.; Anderson, C.; Ko, N.; Deka, R.; Mosley, T.H.; Fornage, M.; Woo, D.; et al. Genome-Wide Association Study of Intracranial Aneurysms Confirms Role of Anril and SOX17 in Disease Risk. Stroke 2012, 43, 2846–2852.
- Wang, X.; Douglas, S.A.; Louden, C.; Vickery-Clark, L.M.; Feuerstein, G.Z.; Ohlstein, E.H. Expression of Endothelin-1, Endothelin-3, Endothelin-Converting Enzyme-1, and Endothelin-A and Endothelin-B Receptor mRNA After Angioplasty-Induced Neointimal Formation in the Rat. Circ. Res. 1996, 78, 322–328.
- Low, S.-K.; Takahashi, A.; Cha, P.-C.; Zembutsu, H.; Kamatani, N.; Kubo, M.; Nakamura, Y. Genome-wide association study for intracranial aneurysm in the Japanese population identifies three candidate susceptible loci and a functional genetic variant at EDNRA. Hum. Mol. Genet. 2012, 21, 2102–2110.
- Yasuno, K.; Bakırcıoğlu, M.; Low, S.-K.; Bilgüvar, K.; Gaál, E.; Ruigrok, Y.M.; Niemelä, M.; Hata, A.; Bijlenga, P.; Kasuya, H.; et al. Common variant near the endothelin receptor type A (EDNRA) gene is associated with intracranial aneurysm risk. Proc. Natl. Acad. Sci. USA 2011, 108, 19707–19712.
- Keedy, A. An overview of intracranial aneurysms. McGill J. Med. MJM Int. Forum Adv. Med. Sci. Stud. 2006, 9, 141–146.
- Krings, T.; Mandell, D.M.; Kiehl, T.-R.; Geibprasert, S.; Tymianski, M.; Alvarez, H.; terBrugge, K.G.; Hans, F.-J. Intracranial aneurysms: From vessel wall pathology to therapeutic approach. Nat. Rev. Neurol. 2011, 7, 547–559.
- Chalouhi, N.; Ali, M.S.; Jabbour, P.M.; Tjoumakaris, S.I.; Gonzalez, L.F.; Rosenwasser, R.H.; Koch, W.J.; Dumont, A.S. Biology of Intracranial Aneurysms: Role of Inflammation. J. Cereb. Blood Flow Metab. 2012, 32, 1659–1676.
- Fennell, V.S.; Kalani, M.Y.S.; Atwal, G.; Martirosyan, N.L.; Spetzler, R.F. Biology of Saccular Cerebral Aneurysms: A Review of Current Understanding and Future Directions. Front. Surg. 2016, 3, 43.
- Tromp, G.; Kuivaniemi, H. Developments in Genomics to Improve Understanding, Diagnosis and Management of Aneurysms and Peripheral Artery Disease. Eur. J. Vasc. Endovasc. Surg. 2009, 38, 676–682.
- Weinsheimer, S.; Lenk, G.M.; Van Der Voet, M.; Land, S.; Ronkainen, A.; Alafuzoff, I.; Kuivaniemi, H.; Tromp, G. Integration of expression profiles and genetic mapping data to identify candidate genes in intracranial aneurysm. Physiol. Genom. 2007, 32, 45–57.
More