Non-Small-Cell Lung Cancer (NSCLC) can harbour different MET alterations, such as MET overexpression (MET OE), MET gene amplification (MET AMP), or MET gene mutations. Retrospective studies of surgical series of patients with MET-dysregulated NSCLC have shown worse clinical outcomes irrespective of the type of specific MET gene alteration. On the other hand, earlier attempts failed to identify the ‘druggable’ molecular gene driver until the discovery of MET exon 14 skipping mutations (METex14). METex14 are rare and amount to around 3% of all NSCLCs. Patients with METex14 NSCLC attain modest results when they are treated with immune checkpoint inhibitors (ICIs). New selective MET inhibitors (MET-Is) showed a long-lasting clinical benefit in patients with METex14 NSCLC and modest activity in patients with MET AMP NSCLC.
- NSCLC
- MET
- MET exon 14 skipping mutations
- MET amplification
- MET overexpression
- MET inhibitors
- prognosis
- immunotherapy
- resistance mechanism
1. Introduction
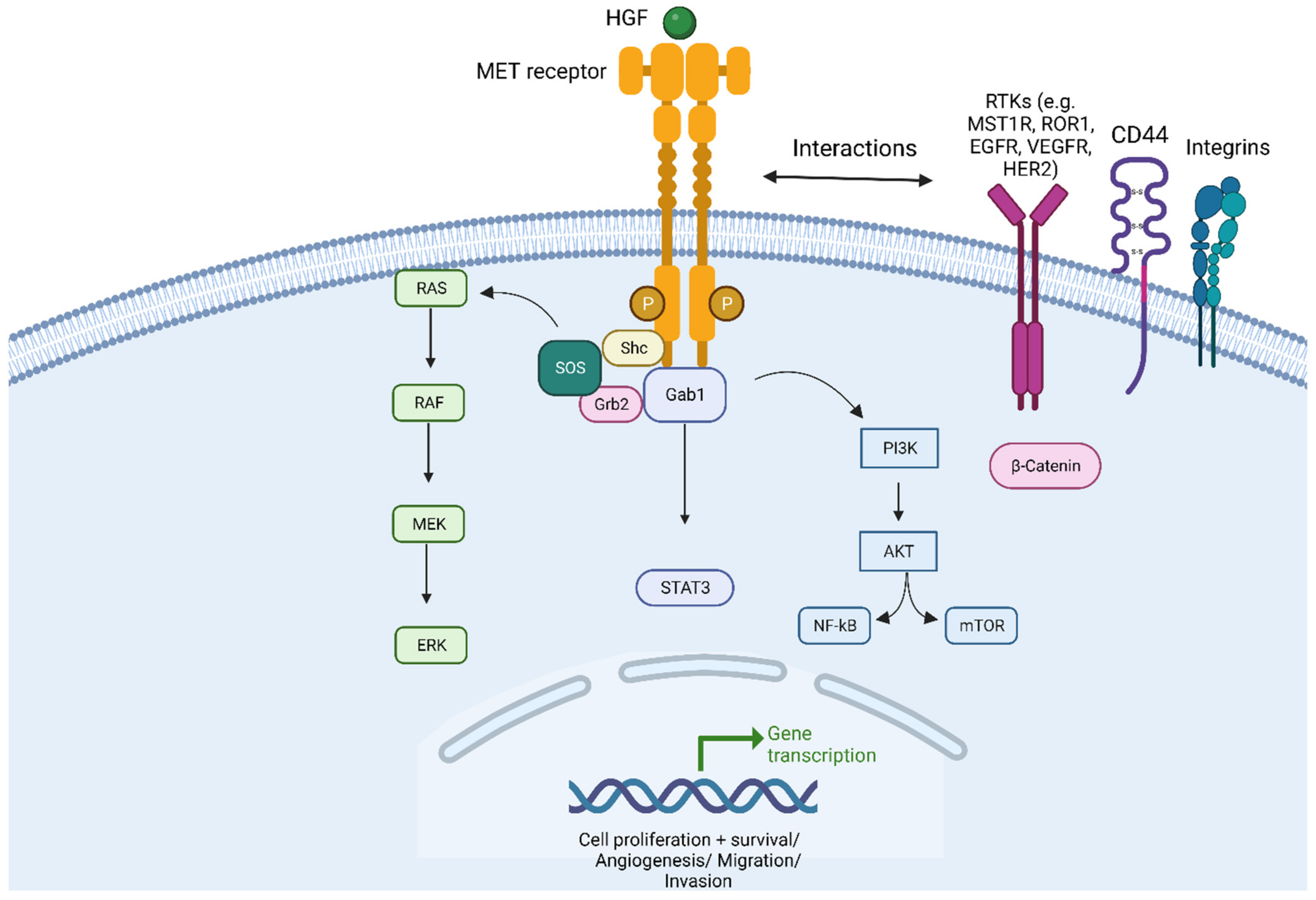
2. Prognostic Role of MET in NSCLC
Some retrospective studies of surgical series have reported the prognostic role of MET dysregulation in NSCLC, which is summarized in Table 1 [18,19,20,21,22,23,24,25,26,27,28,29,30][18][19][20][21][22][23][24][25][26][27][28][29][30].|
Study Reference |
Pt N |
Stage |
Histology |
Methodology |
F-Up (yrs) |
Multivariate Analysis: MET Parameter Correlated with |
|---|---|---|---|---|---|---|
|
Takanami’96 [18] [Japan] |
120 |
I–IV |
ADK |
IHC |
5–12 |
HGF/MET OE: pN and worse OS |
|
Ichimura’96 [19] [Japan] |
104 |
I–IV |
NSCLC |
WB/IHC |
4 |
HGF/MET OE: ADK, p-Stage, worse OS |
|
Siegfried’97 [20] [USA] |
56 |
I–IIIA |
ADK |
HGF IHC |
NR |
HGF OE: worse DFS/OS |
|
Tsao’98 [22] [Canada] |
147 |
I–III |
NSCLC |
mRNA/IHC |
NA |
MET mRNA levels higher in ADK > Sq |
|
Masuya’04 [21] [Japan] |
88 |
I–III |
NSCLC |
HGF/MET IHC |
4.2 |
HGF OE: pT, Ki-67 index, worse OS |
|
Cheng’05 [23] [Taiwan] |
45 |
I–IIIA |
NSCLC |
RT-PCR and IHC |
1.9 |
b-MET mRNA: pN and worse DSF |
|
Nakamura’07 [24] [Japan] |
130 |
I–III |
ADK |
IHC pMET |
2.7 |
Phospho-MET: Grade and papillary |
|
Beau-Faller’08 [25] [France] |
106 |
I–IV |
NSCLC |
GCN RT-PCR |
2.2 |
Higher GCN: worse OS in ADK (trend) |
|
Okuda’08 [26] [Japan] |
213 |
I–IV |
NSCLC |
GCN RT-PCR |
5 * |
Higher GCN: worse OS (for stages II–IV) |
|
Cappuzzo’09 [27] [Italy] |
447 |
I–IV |
NSCLC |
GCN FISH |
3.4 |
Higher GCN: p-Stage, grading, worse OS |
|
Park’12 [28] [Korea] |
380 |
I–IV |
NSCLC |
GCN FISH IHC |
3.5 |
MET OE: worse OS in ADK (trend) |
|
Yeung’15 [29] [Hong Kong] |
154 |
I–IV |
ADK/ADS |
GCN FISH IHC RT-PCR |
2.1 |
METex14: worse OS |
|
Tong’16 [30] [Hong Kong] |
687 |
I–IV |
NSCLC |
FISH IHC RT-PCR |
3.4 |
METex14/High GCN: worse OS |
3. MET as Predictive Biomarker
3.1. MET Exon 14 Alterations
MET exon 14 encodes part of an important regulatory region and a binding site for cBL E3-ubiquitin ligase in the juxtamembrane domain of the MET receptor. Loss of this binding site leads to decreased ubiquitination, thereby reducing MET receptor internalization and degradation, resulting in increased MET levels [31,32,33][31][32][33]. MET ex14 alterations comprised point mutation, insertion, and deletion. Most frequent are disrupted splice sites that lead to MET ex 14 skipping in the RNA transcript and a truncated MET receptor without the Y1003 ubiquitin ligase binding site [34] (Figure 2).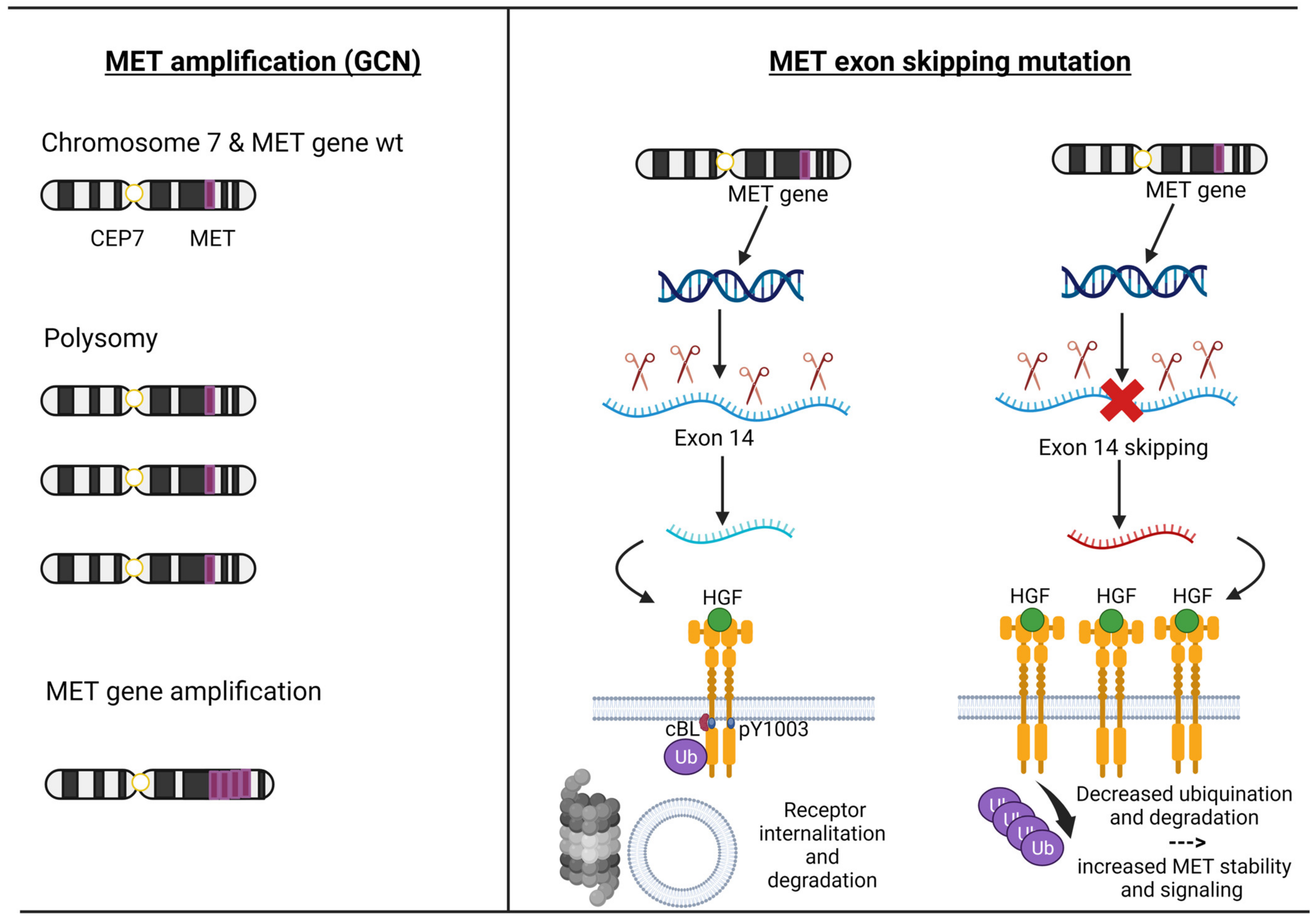
3.2. MET Amplification
The MET amplification (MET AMP) is due to an increase in the copy number of the MET gene (GCN), resulting in protein overexpression. True gene amplification results from gene duplication should not be confused with increased MET copy number due to high polysomy where there is complete chromosome duplication leading to multiple copies of chromosome 7 in tumour cells. Distinguishing between these two mechanisms is critical as true MET amplification is thought to drive oncogenesis, whereas polysomy usually does not [33] (Figure 2). MET amplification (MET AMP) as a ‘druggable driver’ has a longer and more complex history because, in this circumstance, we need to select the real gene-addicted tumours to avoid the ‘dilution effect’ that consists of including a heterogeneity patient population that comprises polysomy and low gene amplification. In fact, the frequency of MET GCN in NSCLC ranges from 0.7% to 21%, depending on the technique used and the cut-off point for positivity [49]. Fluorescence in situ hybridization (FISH) was the first validated method for detecting high MET copy numbers and, to date, remains the gold standard method [48]. One of the first cut-offs used was the University of Colorado Cancer Center (CCC) criteria, which established the gene amplification on the basis of GCNs and the ratio between GCN and centromere of chromosome 7 (MET/CEP7). It also distinguishes low polysomy (≥4 copies in 10–40% of cells), high polysomy (≥4 copies in ≥40% of cells), and gene amplification (presence of tight gene clusters and a MET/CEP7 ratio of ≥2 or 15≥ copies of MET per cell in ≥10% of cells) [50]. The next cut-off was established by Cappuzzo et al., where MET GCN was classified into those with a mean greater than or equal to five copies per cell versus less than five copies. Using this system, they identified 48 (11.1%) patients out of 435 cases, where true amplification was identified in 18 (4.1%) patients [27]. Subsequently, two studies used the Cappuzzo criteria and CCC criteria, finding MET+ cases in 7% and 4.2%, while the true amplification was only 2.4% and 1.1% [28,30][28][30]. Noonan et al. analyzed 1164 patients with adenocarcinoma with information on MET/CEP7 ratio and 686 with information on mean MET per cell value from two separate cohorts, and the GCN was reported by two methods: mean MET copy number per cell value (low ≥ 5 to <6 copies; intermediate ≥ 6 to <7 copies; high ≥ 7 copies) and the ratio MET/CEP7 (low ≥ 1.8 to ≤2.2; intermediate > 2.2 to <5; or high ≥ 5) [51]. Out of 686 cases, 99 (14%) had a MET ≥ 5 copies (FISH+), and 52 of 1164 (4.5%) had a MET/CEP7 ratio ≥ 1.8, meanwhile only 4 (0.03%) patients had a high amplification (ratio ≥ 5). Overall, 61% of 1164 patients had concomitant gene mutations. According to the MET/CEP7 ratio, the concomitant gene mutations were distributed as the following: low (15 of 29, 52%), intermediate (9 of 18, 50%), and high (0 of 4, 0%, p = 0.04). This rigorous cut-off (ratio > 5) helped to identify extremely rare true amplificated cases (less than 1%) without other concomitant gene alterations [51]. However, this analysis was not conclusive, and even now, several cut-offs are being used. It should be noted that MET AMP has been described in patients with NSCLC not previously exposed to tyrosine kinase inhibitors (TKIs) (de novo MET amplification) or as a mechanism of resistance to TKIs, mainly to EGFR (acquired MET amplification).3.3. MET Overexpression
MET protein overexpression ranges from 15 to 70% in NSCLC [28]. The MET receptor overexpression (MET OE) is measured by immunohistochemistry (IHC). It is found that there is a poor correlation with MET gene amplification or mutation. In the Lung Cancer Mutation Consortium multi-institutional cohort study of seventy-one IHC-positive cases, only one (1%) was MET AMP, and two (3%) were METex14-mutated; instead of the 110 MET IHC-negative cases, two (2%) were MET-amplified [52]. In addition, several phase 3 trials with MET overexpression treated with MET-targeted therapies found that protein overexpression was an unreliable biomarker; thus, the utility of this biomarker is limited in clinical practice [52].4. Targeting MET
In the past, two randomized phase III trials with onartuzumab (a monoclonal antibody) and tivantinib (a selective MET TKI inhibitor) in the second/third line were conducted in an unselected NSCLC patient population, and these trials failed to meet their primary endpoint in improving the OS [53,54] (Table S1)[53][54]. Additionally, several MET-Is have been developed despite the failure of these first approaches [53,54][53][54]. MET-Is can be distinguished because of the mechanism of trapping MET: type I inhibitors compete with ATP to the ATP-binding pocket of the active conformation of MET, type Ia (crizotinib) interacts through the G1163 site, while type Ib (capmatinib, tepotinib, and savolitinib) interaction is independent of the G1163 site; type II ATP-competitive MET kinase inhibitors (cabozantinib, glesatinib, and merestinib) are defined by their ability to inhibit MET in its inactive state [55,56][55][56].5. Resistance Mechanisms to MET-Is
5.1. Intrinsic Resistance
It refers to primary resistance and, to outhe researchers' knowledge, is a phenomenon poorly described for MET+ NSCLC treated with MET-I. Moreover, there are currently no translational studies that have investigated epigenetic alterations that could have helped us better understand the mechanisms of resistance to inhibitory METs. Type I MET inhibitors’ activity does not seem to be negatively influenced by the different types or localizations of the METex14 mutations [68,74,84,93][57][58][59][60]. Subgroup analyses for evaluable patients with sufficient tumour samples for NGS from PROFILE-1001 and phase II trial of savolitinib and NGS analyses from liquid biopsies from the VISION trial showed that patients with METex14 NSCLC could have concomitant gene mutations and that the most common are p53 (38–49%), MDM2 (20–25%), and CDKN2A (20%) [68,84,93][57][59][60]. Moreover, post hoc analyses showed that p53 could negatively affect the response (savolitinib trial), and a trend has been documented for the lasting benefit of p53 (VISION and savolitinib trials) and POT-1 in few patients with sarcomatoid histologies (savolitinib trial) [84,93,95][59][60][61]. Importantly, for patients with high MET AMP NSCLC, the absence of concomitant gene mutations was associated with higher ORR in PROFILE-1001 [69][62].5.2. Acquired Resistance
In METex14 or MET AMP NSCLC, patients treated with MET-Is acquired mutations in the tyrosine kinase domain (TKD) and can sustain resistance to these inhibitors. Importantly, D1228 and Y1230 seem to mediate resistance to type I inhibitors by disrupting drug binding, while L1195 and F1200 mutations seem to confer resistance to type II inhibitors [96,97,98,99,100,101,102,103,104][63][64][65][66][67][68][69][70][71]. Moreover, an off-target mechanism of resistance to MET inhibitors involving KRAS gene amplification or mutations has been proved both in preclinical and clinical models [105,106][72][73]. One preclinical study with selected MET-Is showed that mutations within the MET activation loop (D1228N, Y1230C/H) were associated with resistance to type I MET-Is but remained sensitive to type II inhibitors (glesatinib) [98][65]. In a retrospective study, Recondo et al. reported 20 patients with METex14 NSCLC treated with MET-Is [102][69]. They found genomic alterations (GAs) potentially related to resistance to MET-Is in 15 (75%) patients: 7 (35%) patients developed single or compound MET mutations on TKD in codons G1163R, D1228H/N, Y1230C/H/S, and L1195V (associated with type I inhibitors crizotinib and capmatinib) and H1094Y and L1195V (type II inhibitor glesatinib) plus high MET AMP, while 9 (45%) patients were associated with off-target alteration including KRAS (mutations or amplifications) and gene amplification of EGFR, HER3, and BRAF; one patient developed both on- and off-target mechanisms of resistance. It should be noted that two out of six patients (33%) were sensitive to the MET-I switch (the first case was initially treated with crizotinib and subsequently with merestinib; the second was treated with glesatinib and then with crizotinib) [102][69]. A second Chinese retrospective study has been conducted with 86 patients with NSCLC harbouring MET TKD mutations [103][70]. Importantly, they screened three different cohorts of patients (treatment-naïve patients with EGFR+ NSCLC treated with either EGFRTKI and/or MET inhibitors), and they found that ‘acquired’ MET mutations are extremely rare in treatment-naïve patients (0.06%) and have higher frequency after EGFR/MET-Is [103][70]. From a small phase II trial of capmatinib in 20 patients with METex14 NSCLC after crizotinib treatment, among the sixteen patients with detectable ctDNA, five (31%) had acquired MET mutations, three (19%) had MAPK pathway alterations, and two (13%) had ERBB pathway alterations [104][71]. From the VISION trial of tepotinib, 52 pts with progression had end-of-treatment liquid biopsy samples [95][61]. Emerging MET resistance mutations (Y1230H/C and D1228H/N, plus three unknown functions G685E, G344R, and S156L mutations) occurred in seven (13%) patients and off-target mechanisms p53/RB1 mutations (6/35), EGFR/HER2 amplifications (4/35), and PI3K/RAS mutations (3/35) [95][61]. Two independent studies have shown that KRAS alterations (amplification or mutations) can play a significant role in acquired resistance to MET inhibitors [105,106][72][73]. As for MET AMP NSCLC, nine evaluable patients out of twenty-four patients enrolled in cohort B (MET amplification) of the VISION trial have had liquid biopsy profiles at disease progression: two (22.2%) of them had acquired MET resistance mutations; one patient had D1228H/N/Y, Y1230C/H, and D1231N; and another had D1228N/H, Y1230H, and D1231N (90). Figure 3 and Table 2 summarize the intrinsic and acquired mechanisms of resistance to MET-Is.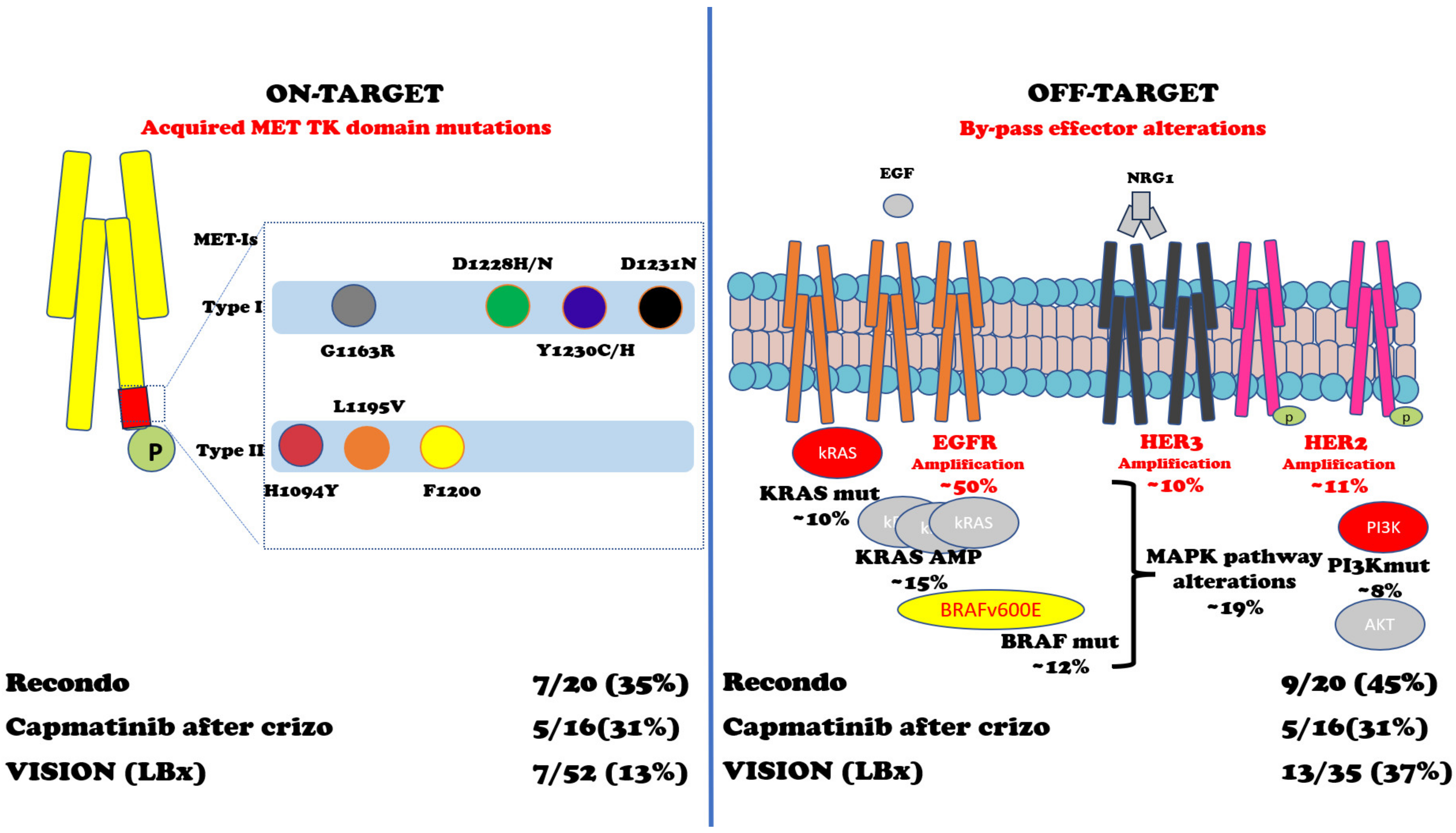
|
Reference |
Study |
Drug |
Type Inh. |
Mechanism of Resistance |
|---|---|---|---|---|
|
On-target |
||||
|
Retrosp. 32/54,752 (0.06%) |
None |
NA |
Extremely rare incidence of MET TK domain mutations in tx-I pts |
|
|
Preclinical |
NVP-BVU972 AMG458 |
I II |
MET Y1230 MET F1200 |
|
|
Preclinical |
MET-Is |
I II |
MET D1228 or Y1230 MET L1195 or F1200 |
|
|
Preclinical |
Type I glesatinib |
I II |
MET1228N or Y1230C/H Sensitive to glesatinib |
|
|
Case report |
Osimertinib+ savolitinib Erlotinib+ cabozantinib |
I II |
EGFR+NSCLC -> MET D1228V Sensitive to cabozantinib |
|
|
Case report |
Crizotinib |
I |
METex14NSCLC -> METD1228N |
|
|
Case report |
Crizotinib |
I |
METex14NSCLC -> Y1230C |
|
|
Retrosp. 7/15 (35%) |
Crizotinib capmatinib Glesatinib |
I II |
G1163R, D1228H/N Y1230 L1195V H1094Y and L1195V |
|
|
Retrosp. 41 pts |
EGFR TKI+ MET (20 pts) MET inh (21 pts) |
NA |
D1228N (63%) D1228H (42%) Y1230H (20%) Y1230C (15%) D1228Y (12%) L1195V (10%) D1228/M1229 (1 pt) |
|
|
Dagogo-Jack |
Phase II 5/16 METex14 (31%) |
Crizotinib |
I |
D1228H (2 patients) Y1230H (1 patient) D1228N/Y1230H (1 patient) |
|
Phase II 7/52 METex14 (13%) |
Tepotinib |
I |
Y1230H/C, D1228H/N plus 3 unknown function G685E, G344R, S156L |
|
|
Phase II 2/9 MET AMP (22%) |
Tepotinib |
I |
D1228H/N/Y, Y1230C/H, D1231N (1) D1228N/H, Y1230H, D1231N (1) |
|
|
Off-target |
||||
|
Preclinical |
Crizotinib |
I |
KRAS gene amplification |
|
|
Retrosp. 1/113 (0.08%) |
Crizotinib |
I |
1/113 post-Crizo KRAS mutation 4/113 pre-treatment KRAS mutations |
|
|
Retrosp. 9/15 METex14 (45%) |
Crizotinib Capmatinib Glesatinib |
I II |
KRAS gene amplification or mutations EGFR gene amplification HER3 gene amplification BRAF gene amplification |
|
|
Dagogo-Jack |
Phase II 5/16 METex14 (31%) |
Crizotinib |
I |
MAPK pathway alterations (3/16) ERBB pathway alterations (2/16) |
|
Phase II 13/35 METex14 (37%) |
Tepotinib |
I |
p53/RB1 mutations (6/35), EGFR/HER2 amplifications (4/35), PI3K/RAS mutations (3/35) |
Abbreviations: MET-Is = MET inhibitors; Type inh = type inhibitors; Retrosp. = retrospective; TK = tyrosine kinase; Tx naïve = treatment-naïve; pts = patients; EGFR+ = Epidermal growth factor receptor mutated; METex14 = MET exon 14 skipping mutations; MET AMP = MET amplification; Crizo = crizotinib; KRAS = Kirsten rat sarcoma virus oncogene; ERBB = EGFR family; HER2 = human epidermal growth factor receptor 2; PI3K = Phosphatidylinositol 3-kinase; MAPK = Mitogen-activated protein kinase.
6. MET and Immunotherapy
Pivotal clinical trials of ICI have excluded or not included patients with known MET alterations NSCLC; therefore, ourthe knowledge of efficacy in this target patient population can only be derived from translational research and retrospective trials [107][75]. MET OE NSCLCs were associated with higher PD-L1 expression and T-cell infiltration [108][76]. MET AMP NSCLC correlated with higher PD-L1 score, CD8+ T-cell infiltration, higher incidence of concomitant gene mutations and worse OS [109][77]. Patients with high MET AMP NSCLC (stated as >10 copies) experienced a statistically significant benefit on OS when treated with ICI added to chemotherapy [110][78]. Instead, there are conflicting results regarding patients with METex14 NSCLC and PD-L1 expression: one study showed that they are associated with higher PD-L1 score [111][79], another study documented higher CD8+ T-cell infiltration but no higher PD-L1 levels [112][80], while another third study found that they did not have high median Tumour mutational burden (mTMB) nor high PD-L1 levels [113][81]. Sabari et al. reported 24 patients with METex14 NSCLC treated with ICI; the ORR was 17%, and the mPFS was 1.9 months. Responses were not enriched in tumours with PD-L1 ≥ 50% nor high TMB [113][81]. Dudnik et al. reported 14 patients with METex14 NSCLC and 5 patients with MET AMP (cut-off not specified) NSCLC [114][82]. The activity of ICI was modest in both groups of patients: in the first, the ORR was 12% (none out of two patients responded to treatment among those with PD-L1 ≥ 50%) with an mPFS of 4 months (1.9 months in the two patients with PD-L1 ≥ 50%); in the latter, the ORR was 25% (one out of four patients included in the analysis) with an mPFS of 4.9 months [114][82]. The IMMUNOTARGET registry was a large European multi-center retrospective trial also involving EGFR and KRAS and enrolled 551 patients with gene-addicted NSCLC [115][83]. ICI was given mostly as the second- or third-line setting. Thirty-six patients had MET+ NSCLC (twenty-three patients with METex14 plus thirteen with MET AMP, cut-off not indicated), the median PD-L1 score was 30, and 76% of the patients were ever smokers. Among these patients, the ORR was 16%, the mPFS (principal endpoint of the study) was 3.4 months, and the OS was 18.4 months. The mPFS was not augmented, regardless of whether we considered the MET alteration subtype, the smoking status, or PD-L1 expression [115][83]. The GFPC01-2018 study has enrolled 30 patients with METex14 NSCLC (enriched for PDL-1 positive and ever smokers) treated with nivolumab or pembrolizumab and showed an ORR of 36%, an mPFS of 4.9 months, and an OS of 13.4 months [116][84]. This was recently reported in two retrospective studies. In the first study of 87 patients with METex14, NSCLC in the mPFS of ICI-based regimens was numerically low (2.4 months in patients with high PD-L1) [117][85]. In the second study of 248 patients with METex14 NSCLC, the mPFS of ICI was inferior with respect to chemotherapy in the first-line setting (229 patients, 3.6 vs. 5 months) and second-line setting (158 patients, 3.3 vs. 3.9 months) [118][86]. Post hoc analyses from GEOMETRY-mono1 and VISION trials have demonstrated that the activity of the MET inhibitor (capmatinib or tepotinib) was irrespective of prior therapies (including ICI); however, the median time to treatment of ICI in the VISION trial was less than 4.5 months, and the OS of patients treated with ICI alone was inferior respect to patients treated with chemotherapy +/− ICI (15.8 vs. 20 months) [78,119][87][88].References
- Montesano, R.; Matsumoto, K.; Nakamura, T.; Orci, L. Identification of fibroblast-derived epithelial morphogen as hepatocyte growth factor. Cell 1991, 61, 901–908.
- Weidner, K.M.; Behrens, J.; Vandekerckhove, J.; Birchmeier, W. Scatter factor: Molecular characteristics and effect on the invasiveness of epithelial cells. J. Cell Biol. 1990, 111, 2097–2108.
- Bussolino, F.; Di Renzo, M.F.; Ziche, M.; Bocchietto, E.; Olivero, M.; Naldini, L.; Gaudino, G.; Tamagnone, L.; Coffer, A.; Comoglio, P.M. Hepatocyte growth factor is a potent angiogenic factor which stimulates endothelial cell motility and growth. J. Cell Biol. 1992, 119, 629–641.
- Brinkmann, V.; Foroutan, H.; Sachs, M.; Weidner, K.M.; Birchmeier, W. Hepatocyte growth factor/scatter factor induces a variety of tissuespecific morphogenic programs in epithelial cells. J. Cell Biol. 1995, 131, 1573–1586.
- Ye, X.; Weinberg, R.A. Epithelial-mesenchymal plasticity: A central regulator of cancer progression. Trends Cell Biol. 2015, 25, 675–686.
- Nakamura, T.; Nawa, K.; Ichihara, A. Partial purification and characterization of hepatocyte growth factor from serum of hepatectomized rats. Biochem. Biophys. Res. Commun. 1984, 122, 1450–1459.
- Stoker, M.; Gherardi, E.; Perryman, M.; Gray, J. Scatter factor is a fibroblast-derived modulator of epithelial cell mobility. Nature 1987, 327, 239–242.
- Comoglio, P.M.; Trusolino, L.; Boccaccio, C. Known and novel roles of the MET oncogene in cancer: A coherent approach to targeted therapy. Nat. Rev. Cancer 2018, 18, 341–358.
- Di Renzo, M.F.; Narsimhan, R.P.; Olivero, M.; Bretti, S.; Giordano, S.; Medico, E.; Gaglia, P.; Zara, P.; Comoglio, P.M. Expression of the Met/HGF receptor in normal and neoplastic human tissues. Oncogene 1991, 6, 1997–2003.
- Olivero, M.; Rizzo, M.; Madeddu, R.; Casadio, C.; Pennacchietti, S.; Nicotra, M.R.; Prat, M.; Maggi, G.; Arena, N.; Natali, P.G.; et al. Overexpression and activation of hepatocyte growth factor/scatter factor in human non-small-cell lung carcinomas. Br. J. Cancer 1996, 74, 1862–1868.
- Rygaard, K.; Nakamura, T.; Spang-Thomsen, M. Expression of the protoncogens c-MET and c-kit and their ligands, hepatocyte growth factor/scatter factor, in SCLC lines and xenografts. Br. J. Cancer 1993, 67, 37–46.
- Comoglio, P.M.; Giordano, S.; Trusolino, L. Drug development of MET inhibitors: Targeting oncogene addiction and expedience. Nat. Rev. Drug Discov. 2008, 7, 504–516.
- Gherardi, E.; Birchmeier, W.; Birchmeier, C.; Vande, W.G. Targeting MET in cancer: Rationale and progress. Nat. Rev. Cancer 2012, 12, 89–103.
- Corso, S.; Migliore, C.; Ghiso, E.; De Rosa, G.; Comoglio, P.M.; Giordano, S. Silencing the MET oncogene leads to regression of experimental tumors and metastases. Oncogene 2008, 27, 684–693.
- Hayward, N.K.; Wilmott, J.S.; Waddell, N.; Johansson, P.A.; Field, M.A.; Nones, K.; Patch, A.-M.; Kakavand, H.; Alexandrov, L.B.; Burke, H.; et al. Whole-genome landscapes of major melanoma subtypes. Nature 2017, 545, 175–180.
- Turke, A.B.; Zejnullahu, K.; Wu, Y.-L.; Song, Y.; Dias-Santagata, D.; Lifshits, E.; Toschi, L.; Rogers, A.; Mok, T.; Sequist, L.; et al. Preexistence and clonal selection of MET amplification in EGFR mutant NSCLC. Cancer Cell 2010, 17, 77–88.
- Kwak, E.L.; Ahronian, L.G.; Siravegna, G.; Mussolin, B.; Borger, D.R.; Godfrey, J.T.; Jessop, N.A.; Clark, J.W.; Blaszkowsky, L.S.; Ryan, D.P.; et al. Molecular Heterogeneity and Receptor Coamplification Drive Resistance to Targeted Therapy in MET-Amplified Esophagogastric Cancer. Cancer Discov. 2015, 5, 1271–1281.
- Takanami, I.; Tanana, F.; Hashizume, T.; Kikuchi, K.; Yamamoto, Y.; Yamamoto, T.; Kodaira, S. Hepatocyte growth factor and c-MET/Hepatocyte growth factor receptor in pulmonary adenocarcinomas: An evaluation of their expression as prognostic markers. Oncology 1996, 53, 392–397.
- Ichimura, E.; Arafumi, M.; Nakajima, T.; Nakamura, T. Expression of c-met/HGF Receptor in Human Non-small Cell Lung Carcinomas in vitro and in vivo and Its Prognostic Significance. Jpn. J. Cancer Res. 1996, 87, 1063–1069.
- Siegfried, J.M.; Weissfeld, L.A.; Singh-Kaw, P.; Weyant, R.J.; Testa, J.R.; Landreneau, R.J. Association of immunoreactive hepatocyte growth factor with poor survival in resectable non-small cell lung cancer. Cancer Res. 1997, 57, 433–439.
- Masuya, D.; Huang, C.; Liu, D.; Kameyama, K.; Haba, R.; Ueno, M.; Yokomise, H. The tumour-stromal interaction between intratumoral c-Met and stromal hepatocyte growth factor associated with tumour growth and prognosis in non-small-cell lung cancer patients. Br. J. Cancer 2004, 90, 1555–1562.
- Tsao, M.S.; Liu, N.; Chen, J.R.; Pappas, J.; Ho, J.; To, C.; Viallet, J.; Park, M.; Zhu, H. Differential expression of Met/hepatocyte growth factor receptor in subtypes of non-small cell lung cancers. Lung Cancer 1998, 20, 1–16.
- Cheng, T.L.; Chang, M.-Y.; Huang, S.-Y.; Sheu, C.-C.; Kao, E.-L.; Cheng, Y.-J.; Chong, I.W. Overexpression of Circulating c-Met Messenger RNA Is Significantly Correlated with Nodal Stage and Early Recurrence in Non-Small Cell Lung Cancer. Chest 2005, 128, 1453–1460.
- Nakamura, Y.; Niki, T.; Goto, A.; Morikawa, T.; Miyazawa, K.; Nakajima, J.; Fukayama, M. c-Met activation in lung adenocarcinoma tissues: An immunohistochemical analysis. Cancer Sci. 2007, 98, 1006–1013.
- Beau-Faller, M.; Ruppert, A.-M.; Voegeli, A.-C.; Neuville, A.; Meyer, N.; Guerin, E.; Legrain, M.; Mennecier, B.; Wihlm, J.-M.; Massard, G.; et al. MET Gene Copy Number in Non-small Cell Lung Cancer: Molecular Analysis in a Targeted Tyrosine Kinase Inhibitor Naïve Cohort. J. Thorac. Oncol. 2008, 3, 331–339.
- Okuda, K.; Sasaki, H.; Yukiue, H.; Yano, M.; Fujii, Y. Met gene copy number predicts the prognosis for completely resected non-small cell lung cancer. Cancer Sci. 2008, 99, 2280–2285.
- Cappuzzo, F.; Marchetti, A.; Skokan, M.; Rossi, E.; Gajapathy, S.; Felicioni, L.; Del Grammastro, M.; Sciarrotta, M.G.; Buttitta, F.; Incarbone, M.; et al. Increased MET Gene Copy Number Negatively Affects Survival of Surgically Resected Non–Small-Cell Lung Cancer Patients. J. Clin. Oncol. 2009, 27, 1667–1674.
- Park, S.; Shin, Y.-L.; Sung, C.O.; An, J.; Seo, J.; Ahn, M.-J.; Ahn, J.S.; Park, K.; Shin, Y.K.; Erkin, O.C.; et al. High MET copy number and MET overexpression: Poor outcome in non-small cell lung cancer patients. Histol. Histopathol. 2012, 27, 197–207.
- Yeung, S.F.; Tong, J.H.M.; Law, P.P.W.; Chung, L.Y.; Lung, R.W.M.; Tong, C.Y.K.; Chow, C.; Chan, A.W.H.; Wan, I.Y.P.; Mok, T.S.K.; et al. Profiling of Oncogenic Driver Events in Lung Adenocarcinoma Revealed MET Mutation as Independent Prognostic Factor. J. Thorac. Oncol. 2015, 10, 1292–1300.
- Tong, J.H.; Yeung, S.F.; Chan, A.W.H.; Chung, L.Y.; Chau, S.L.; Lung, R.W.M.; Tong, C.Y.; Chow, C.; Tin, E.K.Y.; Yu, Y.H.; et al. MET Amplification and Exon 14 Splice Site Mutation Define Unique Molecular Subgroups of Non–Small Cell Lung Carcinoma with Poor Prognosis. Clin. Cancer Res. 2016, 22, 3048–3056.
- Ma, P.C.; Jagadeeswaran, R.; Jagadeesh, S.; Tretiakova, M.S.; Nallasura, V.; Fox, E.A.; Hansen, M.; Schaefer, E.; Naoki, K.; Lader, A.; et al. Functional expression and mutations of c-Met and its therapeutic inhibition with SU11274 and small interfering RNA in non-small cell lung cancer. Cancer Res. 2005, 65, 1479–1488.
- Baek, C.M.; Jeon, S.H.; Jang, J.J.; Lee, B.S.; Lee, J.H. Transforming variant of Met receptor confers serum independence and anti-apoptotic property and could be involved in the mouse thymic lymphomagenesis. Exp. Mol. Med. 2004, 36, 283–291.
- Sakamoto, M.; Patil, T. MET alterations in advanced non-small cell lung cancer. Lung Cancer 2023, 178, 254–268.
- Kong-Beltran, M.; Seshagiri, S.; Zha, J.; Zhu, W.; Bhawe, K.; Mendoza, N.; Holcomb, T.; Pujara, K.; Stinson, J.; Fu, L.; et al. Somatic mutations lead to an oncogenic deletion of met in lung cancer. Cancer Res. 2006, 66, 283–289.
- Frampton, G.M.; Ali, S.M.; Rosenzweig, M.; Chmielecki, J.; Lu, X.; Bauer, T.M.; Akimov, M.; Bufill, J.A.; Lee, C.; Jentz, D.; et al. Activation of MET via diverse exon 14 splicing alterations occurs in multiple tumor types and confers clinical sensitivity to MET inhibitors. Cancer Discov. 2015, 5, 850–859.
- Owad, M.M.; Oxnard, G.R.; Jackman, D.M.; Savukoski, D.O.; Hall, D.; Shivdasani, P.; Heng, J.C.; Dahlberg, S.E.; Jänne, P.A.; Verma, S.; et al. MET Exon 14 Mutations in Non-Small-Cell Lung Cancer Are Associated with Advanced Age and Stage-Dependent MET Genomic Amplification and c-Met Overexpression. J. Clin. Oncol. 2016, 34, 721–730.
- Marks, J.A.; Gandhi, N.; Halmos, B.; Ramalingam, S.S.; Bazhenova, L.; Marmarelis, M.E.; Xiu, J.; Walker, P.; Oberley, M.J.; Ma, P.C.; et al. Updated molecular analysis of MET exon 14skippingmutations (METex14) in non-small cell lung cancer (NSCLC). J. Clin. Oncol. 2023, 41 (Suppl. 16), 9095.
- Socinski, M.A.; Pennell, N.A.; Davies, K.D. MET Exon 14 Skipping Mutations in Non-Small-Cell Lung Cancer: An Overview of Biology, Clinical Outcomes, and Testing Considerations. JCO Precis. Oncol. 2021, 5, PO.20.00516.
- Poirot, B.; Doucet, L.; Benhenda, S.; Champ, J.; Meignin, V.; Lehmann-Che, J. MET exon 14 alterations and new resistance mutations to tyrosine kinase inhibitors: Risk of inadequate detection with current amplicon-based NGS panels. J. Thorac. Oncol. 2017, 12, 1582–1587.
- Descarpentries, C.; Leprêtre, F.; Escande, F.; Kherrouche, Z.; Figeac, M.; Sebda, S.; Baldacci, S.; Grégoire, V.; Jamme, P.; Copinet, M.-C.; et al. Optimization of routine testing for MET exon 14 splice site mutations in NSCLC patients. J. Thorac. Oncol. 2018, 13, 1873–1883.
- Jennings, L.J.; Arcila, M.E.; Corless, C.; Kamel-Reid, S.; Lubin, I.M.; Pfeifer, J.; Temple-Smolkin, R.L.; Voelkerding, K.V.; Nikiforova, M.N. Guidelines for validation of next-generation sequencing-based oncology panels: A joint consensus recommendation of the Association for Molecular Pathology and College of American Pathologists. J. Mol. Diagn. 2017, 19, 341–365.
- Davies, K.D.; Lomboy, A.; Lawrence, C.A.; Yourshaw, M.; Bocsi, G.T.; Camidge, D.R.; Aisner, D.L. DNA-based versus RNA-based detection of MET exon 14 skipping events in lung cancer. J. Thorac. Oncol. 2019, 14, 737–741.
- Jurkiewicz, M.; Saqi, A.; Mansukhani, M.M.; Hodel, V.; Krull, A.; Shu, C.A.; D’Silva Fernandes, H. Efficacy of DNA versus RNA NGS-based methods in MET exon 14 skipping mutation detection. J. Clin. Oncol. 2020, 38 (Suppl. 15), 9036.
- Von Ahlfen, S.; Missel, A.; Bendrat, K.; Schlumpberger, M. Determinants of RNA quality from FFPE samples. PLoS ONE 2007, 2, e1261.
- QIAGEN. Available online: https://www.illumina.com/science/technology/next-generation-sequencing.html (accessed on 6 August 2023).
- Thermofisher Scientific. Available online: https://www.thermofisher.com/it/en/home/life-science/sequencing/next-generation-sequencing.html (accessed on 6 August 2023).
- Sun, R.; Wang, Z.; Zhao, J.; Ren, P.; Ma, J.; Guo, Y. Optimized Detection of Unknown MET Exon 14 Skipping Mutations in Routine Testing for Patients with Non–Small-Cell Lung Cancer. JCO Precis. Oncol. 2023, 7, e2200482.
- Hendriks, L.E.; Kerr, K.M.; Menis, J.; Mok, T.S.; Nestle, U.; Passaro, A.; Peters, S.; Planchard, D.; Smit, E.F.; Solomon, B.J.; et al. Oncogene-addicted metastatic non-small-cell lung cancer: ESMO Clinical Practice Guideline for diagnosis, treatment and follow-up. Ann. Oncol. 2023, 34, 339–357.
- Kris, M.G.; Johnson, B.E.; Berry, L.D.; Kwiatkowski, D.J.; Iafrate, A.J.; Wistuba, I.I.; Varella-Garcia, M.; Franklin, W.A.; Aronson, S.L.; Su, P.F.; et al. Using multiplexed assays of oncogenic drivers in lung cancers to select targeted drugs. JAMA 2014, 311, 1998–2006.
- Hirsch, F.R.; Varella-Garcia, M.; McCoy, J.; West, H.; Xavier, A.C.; Gumerlock, P.; Bunn, P.A., Jr.; Franklin, W.A.; Crowley, J.; Gandara, D.R.; et al. Increased epidermal growth factor receptor gene copy number detected by fluorescence in situ hybridization associates with increased sensitivity to gefitinib in patients with bronchioloalveolar carcinoma subtypes: A Southwest Oncology Group Study. J. Clin. Oncol. 2005, 23, 6838–6845.
- Noonan, S.A.; Berry, L.; Lu, X.; Gao, D.; Barón, A.E.; Chesnut, P.; Sheren, J.; Aisner, D.L.; Merrick, D.; Doebele, R.C.; et al. Identifying the Appropriate FISH Criteria for Defining MET Copy Number–Driven Lung Adenocarcinoma through Oncogene Overlap Analysis. J. Thorac. Oncol. 2016, 11, 1293–1304.
- Guo, R.; Berry, L.D.; Aisner, D.L.; Sheren, J.; Boyle, T.; Bunn, P.A., Jr.; Johnson, B.E.; Kwiatkowski, D.J.; Drilon, A.; Sholl, L.M.; et al. MET IHC Is a Poor Screen for MET Amplification or MET Exon 14 Mutations in Lung Adenocarcinomas: Data from a Tri-Institutional Cohort of the Lung Cancer Mutation Consortium. J. Thorac. Oncol. 2019, 14, 1666–1671.
- Spigel, D.R.; Edelman, M.J.; O’Byrne, K.; Paz-Ares, L.; Mocci, S.; Phan, S.; Shames, D.S.; Smith, D.; Yu, W.; Paton, V.E.; et al. Results from the Phase III trial of Onartuzumab plus Erlotinib versus Erlotinib in previously pretreated stage IIIB or IV Non-Small-Cell Lung Cancer: METLung. J. Clin. Oncol. 2017, 35, 412–420.
- Scagliotti, G.; von Pawel, J.; Novello, S.; Ramlau, R.; Favaretto, A.; Barlesi, F.; Akerley, W.; Orlov, S.; Santoro, A.; Spigel, D.; et al. Phase III Multinational, Randomized, Double-Blind, Placebo-Controlled Study of Tivantinib (ARQ 197) Plus Erlotinib Versus Erlotinib Alone in Previously Treated Patients with Locally Advanced or Metastatic Nonsquamous Non-Small-Cell Lung Cancer. J. Clin. Oncol. 2015, 33, 2667–2674.
- Cui, J.J. Targeting receptor tyrosine kinase MET in cancer: Small molecule inhibitors and clinical progress. J. Med. Chem. 2014, 57, 4427–4453.
- Pargellis, C.; Tong, L.; Churchill, L.; Cirillo, P.F.; Gilmore, T.; Graham, A.G.; Grob, P.M.; Hickey, E.R.; Moss, N.; Pav, S.; et al. Inhibition of p38 MAP kinase by utilizing a novel allosteric binding site. Nat. Struct. Biol. 2002, 9, 268–272.
- Drilon, A.; Clark, J.W.; Weiss, J.; Ou, S.-H.I.; Camidge, D.R.; Solomon, B.J.; Otterson, G.A.; Villaruz, L.C.; Riely, G.J.; Heist, R.S.; et al. Antitumor activity of crizotinib in lung cancers harboring a MET exon 14 alteration. Nat. Med. 2020, 26, 47–51.
- Wolf, J.; Seto, T.; Han, J.-Y.; Reguart, N.; Garon, E.B.; Groen, H.J.M.; Tan, D.S.W.; Hida, T.; de Jonge, M.; Orlov, S.V.; et al. Capmatinib in MET Exon 14–Mutated or MET-Amplified Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2020, 383, 944–957.
- Paik, P.K.; Felip, E.; Veillon, R.; Sakai, H.; Cortot, A.B.; Garassino, M.C.; Mazieres, J.; Viteri, S.; Senellart, H.; Van Meerbeeck, J.; et al. Tepotinib in Non–Small-Cell Lung Cancer with MET Exon 14 Skipping Mutations. N. Engl. J. Med. 2020, 383, 931–943.
- Lu, S.; Fang, J.; Li, X.; Cao, L.; Zhou, J.; Guo, Q.; Liang, Z.; Cheng, Y.; Jiang, L.; Yang, N.; et al. Once-daily savolitinib in Chinese patients with pulmonary sarcomatoid carcinomas and other non-small-cell lung cancers harbouring MET exon 14 skipping alterations: A multicentre, single-arm, open-label, phase 2 study. Lancet Respir. Med. 2021, 9, 1154–1164.
- Paik, P.K.; Veillon, R.; Felip, E.; Cortot, A.; Sakai, H.; Mazieres, J.; Thomas, M.; Reinmuth, N.; Raskin, J.; Conte, P.F.; et al. METex14 ctDNA dynamics & resistance mechanisms detected in liquid biopsy (LBx) from patients (pts) with METex14 skipping NSCLC treated with tepotinib. J. Clin. Oncol. 2021, 39 (Suppl. 15), 9012.
- Camidge, D.R.; Otterson, G.A.; Clark, J.W.; Ou, S.-H.I.; Weiss, J.; Ades, S.; Shapiro, G.I.; Socinski, M.A.; Murphy, D.A.; Conte, U.; et al. Crizotinib in Patients With MET-Amplified NSCLC. J. Thorac. Oncol. 2021, 16, 1017–1029.
- Tiedt, R.; Degenkolbe, E.; Furet, P.; Appleton, B.A.; Wagner, S.; Schoepfer, J.; Buck, E.; Ruddy, D.A.; Monahan, J.E.; Jones, M.D.; et al. A drug resistance screen using a selective MET inhibitor reveals a spectrum of mutations that partially overlap with activating mutations found in cancer patients. Cancer Res. 2011, 71, 5255–5264.
- Fujino, T.; Kobayashi, Y.; Suda, K.; Koga, T.; Nishino, M.; Ohara, S.; Chiba, M.; Shimoji, M.; Tomizawa, K.; Takemoto, T.; et al. Sensitivity and resistance of MET exon 14 mutations in lung cancer to eight MET tyrosine kinase inhibitors in vitro. J. Thorac. Oncol. 2019, 14, 1753–1765.
- Engstrom, L.D.; Aranda, R.; Lee, M.; Tovar, E.A.; Essenburg, C.J.; Madaj, Z.; Chiang, H.; Briere, D.; Hallin, J.; Lopez-Casas, P.P.; et al. Glesatinib exhibits antitumor activity in lung cancer models and patients harboring MET Exon 14 mutations and overcomes mutation-mediated resistance to type I MET inhibitors in nonclinical models. Clin. Cancer Res. 2017, 23, 6661–6672.
- Bahcall, M.; Sim, T.; Paweletz, C.P.; Patel, J.D.; Alden, R.S.; Kuang, Y.; Sacher, A.G.; Kim, N.D.; Lydon, C.A.; Awad, M.M.; et al. Acquired METD1228V mutation and resistance to MET inhibition in lung cancer. Cancer Discov. 2016, 6, 1334–1341.
- Ou, S.-H.I.; Young, L.; Schrock, A.B.; Johnson, A.; Klempner, S.J.; Zhu, V.W.; Miller, V.A.; Ali, S.M. Emergence of Preexisting MET Y1230C Mutation as a Resistance Mechanism to Crizotinib in NSCLC with MET Exon 14 Skipping. J. Thorac. Oncol. 2017, 12, 137–140.
- Heist, R.S.; Sequist, L.V.; Borger, D.; Gainor, J.F.; Arellano, R.S.; Le, L.P.; Dias-Santagata, D.; Clark, J.W.; Engelman, J.A.; Shaw, A.T.; et al. Acquired resistance to crizotinib in NSCLC with MET exon 14 skipping. J. Thorac. Oncol. 2016, 11, 1242–1245.
- Recondo, G.; Bahcall, M.; Spurr, L.F.; Che, J.; Ricciuti, B.; Leonardi, G.C.; Lo, Y.-C.; Li, Y.Y.; Lamberti, G.; Nguyen, T.; et al. Molecular Mechanisms of Acquired Resistance to MET Tyrosine Kinase Inhibitors in Patients with MET Exon 14–Mutant NSCLC. Clin. Cancer Res. 2020, 26, 2615–2625.
- Yao, Y.; Yang, H.; Zhu, B.; Wang, S.; Pang, J.; Wu, X.; Xu, Y.; Zhang, J.; Zhang, J.; Ou, Q.; et al. Mutations in the MET tyrosine kinase domain and resistance to tyrosine kinase inhibitors in non-small-cell lung cancer. Respir. Res. 2023, 24, 28.
- Dagogo-Jack, I.; Moonsamy, P.; Gainor, J.F.; Lennerz, J.K.; Piotrowska, Z.; Lin, J.J.; Lennes, I.T.; Sequist, L.V.; Shaw, A.T.; Goodwin, K.; et al. A Phase 2 Study of Capmatinib in Patients With MET-Altered Lung Cancer Previously Treated with a MET Inhibitor. J. Thorac. Oncol. 2021, 16, 850–859.
- Bahcall, M.; Awad, M.M.; Sholl, L.M.; Wilson, F.H.; Xu, M.; Wang, S.; Palakurthi, S.; Choi, J.; Ivanova, E.V.; Leonardi, G.C.; et al. Amplification of wild-type KRAS imparts resistance to crizotinib in MET exon 14 mutant non small cell lung cancer. Clin. Cancer Res. 2018, 24, 5963–5976.
- Suzawa, K.; Offin, M.; Lu, D.; Kurzatkowski, C.; Vojnic, M.; Smith, R.S.; Sabari, J.K.; Tai, H.; Mattar, M.; Khodos, I.; et al. Activation of KRAS mediates resistance to targeted therapy in MET exon 14 mutant non small cell lung cancer. Clin. Cancer Res. 2018, 25, 1248–1260.
- Le, X.; Paz-Ares, L.G.; Van Meerbeeck, J.; Ramirez, S.V.; Galvez, C.C.; Baz, D.V.; Kim, Y.-C.; Kang, J.-H.; Stroh, C.; Juraevaet, D.; et al. Clinical response to tepotinib according to circulating tumor (ct) DNA biomarkers in patients with advanced NSCLC with high-level MET amplification (METamp) detected by liquid biopsy (LBx). J. Clin. Oncol. 2022, 40 (Suppl. 16), 9121.
- Vokes, N.I.; Pan, K.; Le, X. Efficacy of immunotherapy in oncogene-driven non-small-cell lung cancer. Ther. Adv. Med. Oncol. 2023, 15, 17588359231161409.
- Yoshimura, K.; Inoue, Y.; Tsuchiya, K.; Karayama, M.; Yamada, H.; Iwashita, Y.; Kawase, A.; Tanahashi, M.; Ogawa, H.; Inui, N.; et al. Elucidation of the relationships of MET protein expression and gene copy number status with PD-L1 expression and the immune microenvironment in non-small cell lung cancer. Lung Cancer 2020, 141, 21–31.
- Yoshimura, K.; Inoue, Y.; Inui, N.; Karayama, M.; Yasui, H.; Hozumi, H.; Suzuki, Y.; Furuhashi, K.; Fujisawa, T.; Enomoto, N.; et al. MET Amplification and Efficacy of Nivolumab in Patients With NSCLC. JTO Clin. Res. Rep. 2021, 2, 100239.
- Kron, A.; Scheffler, M.; Heydt, C.; Ruge, L.; Schaepers, C.; Eisert, A.-K.; Merkelbach-Bruse, S.; Riedel, R.; Nogova, L.; Fischer, R.N.; et al. Genetic Heterogeneity of MET-Aberrant NSCLC and Its Impact on the Outcome of Immunotherapy. J. Thorac. Oncol. 2021, 16, 572–582.
- Schoenfeld, A.J.; Rizvi, H.; Bandlamud, C.; Sauter, J.L.; Travis, W.D.; Rekhtman, N.; Plodkowski, A.J.; Perez-Johnston, R.; Sawan, P.; Beras, A.; et al. Clinical and molecular correlates of PD-L1 expression in patients with lung adenocarcinomas. Ann. Oncol. 2020, 31, 599–608.
- Li, X.; Wang, R.; Wang, L. MET-mutant cancer and immune checkpoint inhibitors: A large database analysis. Lung Cancer 2020, 150, 256–258.
- Sabari, J.K.; Leonardi, G.C.; Shu, C.A.; Umeton, R.; Montecalvo, J.; Ni, A.; Chen, R.; Dienstag, J.; Mrad, C.; Bergagnini, I.; et al. PD-L1 expression, tumor mutational burden, and response to immunotherapy in patients with MET exon 14 altered lung cancers. Ann. Oncol. 2018, 29, 2085–2091.
- Dudnik, E.; Bsharab, E.; Grubsteinc, A.; Fridel, L.; Shochat, T.; Roisman, L.C.; Ilouze, M.; Rozenblum, A.B.; Geva, S.; Zer, A.; et al. Rare targetable drivers (RTDs) in non-small cell lung cancer (NSCLC): Outcomes with immune check-point inhibitors (ICPi). Lung Cancer 2018, 124, 117–124.
- Mazieres, J.; Drilon, A.; Lusque, A.; Mhanna, L.; Cortot, A.B.; Mezquita, L.; Thai, A.A.; Mascaux, C.; Couraud, S.; Veillon, R.; et al. Immunecheckpoint inhibitors for patients with advanced lung cancer and oncogenic driver alterations: Results from the IMMUNOTARGET registry. Ann. Oncol. 2019, 30, 1321–1328.
- Guisier, F.; Dubos-Arvis, C.; Viñas, F.; Doubre, H.; Ricordel, C.; Ropert, S.; Janicot, H.; Bernardi, M.; Fournel, P.; Lamy, R.; et al. Efficacy and safety of anti–PD-1 immunotherapy in patients with advanced NSCLC with BRAF, HER2, or MET mutations or RET translocation: GFPC01-2018. J. Thorac. Oncol. 2020, 15, 628–636.
- Leighl, N.N.; Hampe, M.; Wu, W.-H.; Kim, J.; Pretre, V.; Ye, F. Real-world treatment (tx) patterns and outcomes based on PD-L1 status in tx-naive patients (pts) with METex14 advanced non-small cell lung cancer (aNSCLC). Ann. Oncol. 2022, 33, S1059–S1060.
- Ho, C.; Wong, S.; Hatswell, A.; Slater, R.; Vioix, H.; Chouaid, C. Treatment patterns and progression-free survival in MET exon 14 (METex14) skipping advanced non-small cell lung cancer (aNSCLC) in real-world clinical practice. Ann. Oncol. 2022, 33, S1087–S1088.
- Vansteenkiste, J.; Smit, E.F.; Groen, H.J.M.; Doban, V.; Kanakamedala, H.; Wu, W.; Joshi, A.; de Jong, E.; Giovannini, M.; Baik, C.S. Capmatinib in Patients with METex14-Mutated Advanced NSCLC Who Received Prior Immunotherapy: Results from the Phase 2 GEOMETRY Mono-1 Study. Ann. Oncol. 2020, 31 (Suppl. 4), S754–S840.
- Griesinger, F.; Felip, E.; Smit, E.F.; Veillon, R.; Raskin, J.; Thomas, M.; Conte, P.; Kowalski, D.; Paz-Ares, L.; Garcia Ledo, G.; et al. Tepotinib in patients with MET exon 14 skipping NSCLC: Efficacy and safety by line of therapy. Ann. Oncol. 2022, 33, S40–S41.