Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Ashraf Abadi and Version 2 by Sirius Huang.
Phosphodiesterase 5 (PDE5) inhibitors presented themselves as important players in the nitric oxide/cGMP pathway, thus exerting a profound impact on various physiological and pathological processes. Beyond their well-known efficacy in treating male erectile dysfunction (ED) and pulmonary arterial hypertension (PAH), a plethora of studies have unveiled their significance in the treatment of a myriad of other diseases, including cognitive functions, heart failure, multiple drug resistance in cancer therapy, immune diseases, systemic sclerosis and others.
- phosphodiesterase 5 inhibitors
- selectivity
- NO/cGMP
1. Introduction
PDE5 inhibitors (PDE5-Is) are groundbreaking medications for treating ED. They increase cGMP levels, causing muscle relaxation and vasodilation in the penis, leading to erections. Their therapeutic potential extends beyond ED, with clinical approval for treating PAH, BPH, and LUTS. Research highlights their potential in diseases like cancer, neurological disorders, cystic fibrosis, and diabetes. Promisingly, this has incited the development of new PDE5-Is with higher potency, selectivity, and improved pharmacokinetics for enhanced efficacy.
2. Classification of Phosphodiesterases
PDE superfamily comprises 11 families (PDE1–PDE11) that are encoded by 21 different genes, whose expression are modulated via multiple promotors and messenger RNA (mRNA) alternative splicing generating more than 50 isoforms [1][2][1,2]. It is worth noting that PDE12, which cleaves 2′,5′-phosphodiester bond linking adenosines of the 5′-triphosphorylated oligoadenylates, belongs to the C–C chemokine receptor 4 (CCR4)/nocturin family [3] and is not a member of the cyclic nucleotide PDE superfamily. PDE isoforms are classified based on their amino acid sequences, substrate specificities, catalytic and cofactor requirements, kinetic properties, regulatory mechanisms, and tissue distributions [1]. Some PDEs are selective for the hydrolysis of cAMP (PDE 4, 7, and 8) or cGMP (PDE 5, 6 and 9), while others can hydrolyze both cAMP and cGMP (PDE 1, 2, 3, 10 and 11) [4]. PDEs share a conserved catalytic domain (C domain) but differ significantly in their N-terminal regulatory domains. PDEs are mainly regulated via (i) binding of Ca2+/calmodulin (PDE1), (ii) phosphorylation/dephosphorylation events (PDE1, 3, 4 and 5) and (iii) allosteric binding of cGMP via GAF domains (PDE2, 5, 6, 10 and 11) [1]. The description of diverse tissue distribution/cell expression and functional significance of PDE isoenzymes is detailed in [5] and is beyond the scope of this discussion. Notably, such tissue/cellular compartmentalization allows selective PDE inhibitors to exert their effects almost exclusively on the target tissue. The focus herein is on the PDE5 family, which is generated by one gene, PDE5A, and has three alternative spliced variants, PDE5A1, 5A2 and 5A3. The three human PDE5 isoforms differ only in the 5’-end of the mRNA and the corresponding N-terminal of the protein. These isoforms have similar phosphorylation sites, allosteric cGMP-binding sites, catalytic domain and cGMP binding and hydrolysis activities [6]. However, PDE5A1 was reported to be more resistant to chemical inhibition than PDE5A2 or PDE5A3. PDE5A1 and PDE5A2 are widely distributed in nearly all tissues, whereas PDE5A3 is confined to vascular smooth muscle cells [2].3. Tissues and Organs of High Expression for PDE5
PDE5 is present in virtually all cell types, tissues and organs. PDE5 is highly expressed in the smooth muscle cells of the peripheral arteries and venous vessels and in coronary and pulmonary arteries [2]. In addition, PDE5 is expressed in the vascular smooth muscle cells of the corpora cavernosa of the penis besides spermatozoa, peritubular myoid of Leydig cells and vas deferens in males [7]. PDE5 is widely distributed in the cytoplasmic cell compartment in myometrial cells, endothelial cells and peripheral blood mononuclear cells. It is also expressed in skeletal muscles, cardiomyocytes, platelets, lung, spinal cord, cerebellum, retina, pancreas, prostate, urethra and bladder [1][2][8][1,2,8]. PDE5A1 and PDE5A2 are further expressed in renal vessels, glomeruli, tubular epithelial cells of the renal proximal tubule and medullary collecting duct [9]. Consequently, PDE5 isoforms exhibit diverse and numerous functions both in physiological and pathological conditions.4. PDE5 Physiological Role
Nitric oxide (NO) is synthesized from the precursor L-arginine through the activities of different NO synthases (neuronal, inducible or endothelial NOS). Intracellularly, NO binds to and activates soluble guanylyl cyclase (sGC), promoting the conversion of guanosine triphosphate (GTP) to the second messenger cyclic guanosine monophosphate (cGMP) [10][11][10,11]. Thereafter, cGMP activates protein kinase G (PKG), whose phosphorylation mediates activities of various membrane channels/pumps, leading to decreased calcium influx through L-type calcium channels and increased calcium sequestration, resulting in smooth muscle relaxation and vascular tone modulation [12]. PKG-dependent phosphorylation of other various downstream proteins can regulate further pivotal physiological functions, such as cell differentiation and proliferation, endothelial permeability, ion transport, secretion and gene transcription [13]. Given the broad expression and the ability of PDE5 to specifically hydrolyze cGMP, controlling its cellular levels, PDE5 has been proposed as a crucial player in many NO/cGMP/PKG-dependent biological processes such as smooth muscle relaxation, heart muscle contraction, platelet activation/aggregation and immune response [14]. PDE5 inhibition was found to enhance smooth muscle relaxation and vasodilation, which in the penis corpus cavernosum favors erection, in the pulmonary vasculature decreases pulmonary vessels’ pressure, and in the systemic circulation decreases arterial blood pressure [15]. In addition, PDE5 is an important regulator of platelet function, whose inhibition increases platelet cGMP levels and augments the ability of NO to inhibit platelet aggregation and activation [16]. Furthermore, PDE5 governs fundamental physiological processes in the kidney. It can regulate renal vascular blood flow by hampering cGMP-mediated vascular relaxation. PDE5 is also a negative regulator for cGMP-dependent natriuresis. Moreover, it increases renin synthesis by degrading cGMP in juxtaglomerular cells [17].5. PDE5 as a Drug Target for Disease Treatment
Competitive PDE5-Is reported so far exclusively bind to the catalytic domain, preventing cGMP hydrolysis and elevating its levels in cells of various tissues [18]. The subsequent activation or restoration of normal NO/cGMP/PKG signaling cascade prompted the use of these inhibitors as therapeutics for several clinical indications. Food and Drug Administration (FDA)-approved PDE5-Is (Figure 1) include (i) sildenafil (approved in 1998 for erectile dysfunction (ED) as Viagra®, and in 2005 for pulmonary arterial hypertension (PAH, WHO Group I) as Revatio®), (ii) vardenafil (approved in 2003 for ED as Levitra®), (iii) tadalafil (approved in 2003 for ED as Cialis®, in 2009 for PAH (WHO Group I) as Adcirca® and in 2011 for lower urinary tract symptoms secondary to benign prostatic hyperplasia (LUTS/BPH) with or without ED and the most recent (iv) avanafil (approved in 2012 for ED as Stendra®) [19][20][19,20].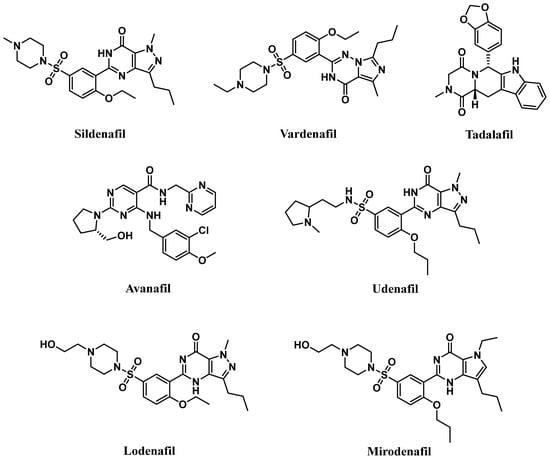
Figure 1.
Chemical structures of marketed PDE5 inhibitors.
5.1. Approved Clinical Uses of PDE5 Inhibitors
5.1.1. Erectile Dysfunction
In the corpora cavernosa, parasympathetic stimulation and sexual arousal induce the release of NO from endothelial cells and nitrergic neurons surrounding the arteries and sinusoids, leading to increased cGMP synthesis. PDE5-Is can slow the degradation of penile connective tissue cGMP. This leads to a drop in the intracellular Ca2+ levels in the corpus cavernosum smooth muscles, causing their relaxation and a reduction in arterial blood drainage, providing a sufficient degree of penile tumescence and sustaining penile erection (Figure 2). Accordingly, it can be deduced that the action of PDE5-Is requires normal neuronal input into the erectile tissues, as well as unimpaired cavernous endothelial structures [24][25][24,25].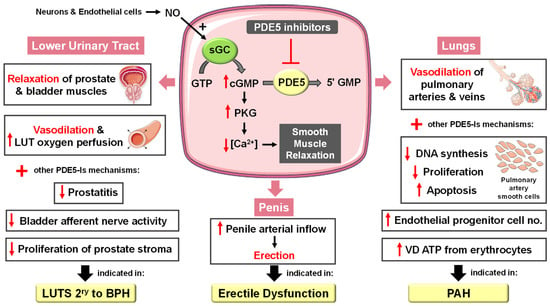
Figure 2. Approved clinical uses of PDE5 inhibitors. Nitric oxide (NO) is produced by neurons and endothelial cells. Inside smooth muscle cells, NO activates soluble guanylyl cyclase (sGC), promoting the conversion of guanosine triphosphate (GTP) to the second messenger cyclic guanosine monophosphate (cGMP). Thereafter, cGMP activates protein kinase G (PKG), whose phosphorylation mediates activities of various membrane channels/pumps, leading to decreased intracellular calcium levels resulting in smooth muscle relaxation (SMR). Phosphodiesterase 5 (PDE5) regulates cGMP levels by degrading it into inactive 5′ guanosine monophosphate (5′ GMP). PDE5-Is can thus enhance the cGMP/PKG pathway, boosting the relaxation of various smooth muscles. In the penis corpus cavernosum, SMR favors erection due to increased penile arterial inflow, and thus PDE5-Is are approved for the treatment of erectile dysfunction. In the lungs, PDE5-Is lead to vasodilation of pulmonary vasculature, which, along with other mechanisms, such as suppressed DNA synthesis and proliferation and enhanced apoptosis of pulmonary artery cells, increased endothelial progenitor cell number, and enhanced release of vasodilating adenosine triphosphate (ATP) from erythrocytes culminate in effectiveness in the treatment of pulmonary arterial hypertension (PAH). In the lower urinary tract (LUT), PDE5-Is mediate prostate and bladder SMR, vasodilation and increased LUT oxygen perfusion. In addition, PDE5-Is could suppress prostatitis, bladder afferent nerve activity and prostate stroma cell proliferation, and thus indicated in the treatment of LUT symptoms secondary to benign prostatic hyperplasia (BPH).
5.1.2. Pulmonary Arterial Hypertension
PAH is a disease associated with endothelial dysfunction, vascular remodeling and fibrosis that causes gradual progression of pulmonary vascular resistance, ultimately leading to right heart failure. Accordingly, PAH therapies usually aim to enhance vasodilation, suppress cellular hyperproliferation and induce apoptosis [33]. PDE5 is highly expressed in the lung vasculature [34]. The fact that lung endothelial NOS is reduced [35] and PDE5 is upregulated in the remodeled pulmonary artery during PAH has proposed PDE5-Is as a potential PAH treatment [36]. A plethora of PDE5 inhibition-mediated mechanisms have been documented (Figure 2) including (i) activation of the NO/cGMP/PKG pathway, resulting in decreased calcium influx through L-type calcium channels and increased calcium sequestration, inducing vasorelaxation [37], (ii) suppression of DNA synthesis and cell proliferation and stimulation of apoptosis of pulmonary artery smooth cells whose proliferation is involved in the pathogenesis of intimal hyperplasia and major vascular lesions in PAH [38], and (iii) increasing circulating endothelial progenitor cell (EPC) number [39]. Several clinical studies confirmed the potential of PDE5-Is to improve several hemodynamic and clinical parameters in PAH patients [40][41][42][43][40,41,42,43], such as diminishing pulmonary artery systolic and mean artery pressure, dyspnea score and gas transfer, pulmonary vascular resistance and cardiac output [44]. Furthermore, PDE5-Is could improve ventilatory efficiency and oxygen uptake kinetics and prevent exercise-induced pulmonary edema [45]. Vardenafil usually exhibits the most rapid effect on pulmonary vasorelaxation, while sildenafil and tadalafil are more selective for pulmonary circulation. Substantial enhancement of arterial oxygenation is mainly observed with sildenafil [46]. Sildenafil, in 2005, and, thereafter, tadalafil have been FDA approved and became first-line therapies for PAH [47], primary or secondary to other connective tissue diseases, such as scleroderma (SSc) or systemic lupus erythematosus (SLE) [34]. PDE5-Is can also be used as combination therapy with other PAH-targeted treatments. The combination of sildenafil and long-term intravenous epoprostenol therapy was superior to epoprostenol monotherapy regarding improved exercise capacity, hemodynamic measurements and prolonged time to clinical worsening [48]. Other combinations, such as tadalafil with the endothelin receptor antagonist ambrisentan and sildenafil with systemic nitrates [49], were proven safe and effective in potentiating vasodilation and reducing mortality in PAH patients. Moreover, combined prostacyclin analogs and PDE5-Is were reported to synergistically enhance the release of the potent vasodilator ATP from PAH erythrocytes [50]. PDE5 inhibition has also emerged as a therapeutic strategy for high-altitude PAH where sildenafil’s ability to reverse hypoxia-mediated pulmonary vasoconstriction was proved to mediate positive results on exercise performance and lung hemodynamics [51][52][51,52].5.1.3. Lower Urinary Tract Symptoms Secondary to Benign Prostatic Hyperplasia
Several studies have established an association between ED and BPH-related LUTS where alterations in the NO/cGMP pathway, alterations in RhoA/Rho kinase/endothelin signaling, pelvic atherosclerosis, autonomic adrenergic hyperactivity, inflammatory pathways, sex hormones and psychological factors were the major contributing factors [53][54][53,54]. Accordingly, attention was drawn towards the development of a single therapy to treat both conditions. The clinical benefits of chronic PDE5 inhibition on LUTS secondary to BPH, regardless of whether these symptoms are associated with ED, are well documented [55]. These beneficial effects have been correlated to several mechanisms (Figure 2), including (i) stromal smooth muscle relaxation of the prostate and bladder due to modulation of the NO/cGMP pathway in the nitrinergic innervated organs or enhanced generation of relaxing hydrogen sulfide, (ii) significant cGMP-mediated dilatation of local blood vessels, (iii) enhanced LUT oxygen perfusion, (iv) inhibition of afferent nerve activity of bladder, (v) down-regulation of prostate inflammation and (vi) negative regulation of proliferation and trans-differentiation of the prostate stroma [54][56][57][54,56,57]. Many preclinical studies of PDE5 and its inhibitors in the prostate and bladder (reviewed in [58]) could validate the role of PDE5-Is in relaxing prostatic tissue, improving the severity of urinary symptoms, reducing bladder overactivity, decreasing indicators of bladder ischemia, normalizing changes in NOS activity and preventing the accumulation of collagen [59]. Several clinical trials demonstrated that the use of PDE5-Is alone could ameliorate LUTS in the first 12 weeks of treatment, where sildenafil [60], tadalafil [61][62][63][61,62,63] and vardenafil [64] led to a decrease, at different degrees, in the International Prostate Symptom Score (IPPS) scale. In particular, the effects of tadalafil 5 mg once daily versus placebo on LUTS/BPH have been extensively investigated (reviewed by Gacci et al. [65]). Only tadalafil (5 mg once daily) has been licensed for the treatment of LUTS with or without ED. The combined administration of sildenafil, tadalafil or vardenafil with the α1-adrenoceptor antagonists alfuzosin or tamsulosin for the treatment of LUTS/BPH has also been evaluated and was confirmed to often outperform either type of monotherapy [66][67][68][69][70][66,67,68,69,70]. Interestingly, a very recent meta-analysis of randomized clinical trials demonstrated that tadalafil could be superior to tamsulosin in treating LUTS/BPH when associated with ED [71].5.2. Emerging and Future Uses of PDE5 Inhibitors
5.2.1. Cancer
Numerous studies have reported the role of cGMP in suppressing cell growth and inducing apoptosis and that elevated PDE5 expression is involved in the progression of various tumor types, such as chronic lymphocytic leukemia, colon adenocarcinoma, bladder squamous carcinoma, human papillary thyroid carcinomas, metastatic breast, prostate, pancreatic and lung cancers [72][73][74][72,73,74]. Accordingly, PDE5 has gained attention as a promising target for anticancer drug discovery. Over the last two decades, several pre-clinical and clinical studies revealed potential anti-cancer effects of PDE5-Is [75][76][75,76] that were mediated via different mechanisms of action discussed herein (Figure 3).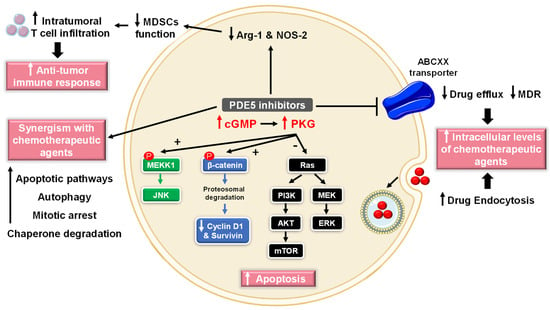
Figure 3. Anti-cancer mechanisms of PDE5 inhibitors. Via activation of the cGMP/PKG signaling cascade, PDE5-Is can induce apoptosis in cancer cells via various pathways; activation of c-Jun NH2-terminal kinase (JNK) via phosphorylation of mitogen-activated protein kinase kinase kinase 1 (MEKK1), phosphorylation of β-catenin and inducing its proteosomal degradation which leads to decreased expression of Wnt/β-catenin regulated proteins, such as cyclin D1 and survivin in addition to blocking the phosphoinositide 3-kinase (PI3K)/AKT/mammalian target of rapamycin (mTOR) and the mitogen-activated protein kinase kinase/extracellular signal-regulated kinase (MEK/ERK) signaling pathways. PDE5-Is could also increase intracellular levels of other chemotherapeutic agents via inhibition of the ATP-binding cassette (ABC) transporter-mediated drug efflux, averting multidrug resistance (MDR) in addition to increasing cellular drug uptake via enhancing endocytosis. Moreover, PDE5-Is synergize with other chemotherapeutic agents via boosting various apoptotic, autophagy, mitotic arrest and chaperone degradation pathways. PDE5-Is can also abrogate the function of myeloid-derived suppressor cells (MDSCs) via suppression of arginase-1 (Arg-1) and nitric oxide synthase–2 (NOS-2) production. This results in enhanced intratumoral T-cell infiltration and activation and restores both systemic and tumor-specific immunity. P = phosphorylation.
- (1)
-
Cell growth arrest and induction of apoptosis
- (2)
-
Chemotherapy sensitization
- (3)
-
Modulation of antitumor immune response
- (4)
-
Chemopreventive mechanisms
5.2.2. CNS Diseases
cGMP/PKG signaling has been regarded as a central mechanism of neuroinflammation, neurodegeneration and cognitive disorders [106][107][106,107]. Accordingly, PDE5-Is have gained growing attention as potential therapeutic agents for the treatment of several CNS-related diseases, such as Alzheimer’s disease (AD), cognitive deficits, strokes, multiple sclerosis (MS), depression, noise-induced hearing loss (NIHL) and neuropathic pain that will all be discussed in this section (Figure 4).PDE5 inhibition increases presynaptic cGMP levels, which, through PKG activation, enhances the release of glutamate and activates N-methyl-D-aspartate (NMDA) receptors. On the other hand, postsynaptic PKG activates transcription factor cyclic adenosine monophosphate (cAMP) response element-binding element (CREB), promoting neurotransmission, synaptic plasticity and memory consolidation [108][109][108,109]. PKG also activates the PI3K/AKT signaling pathway that mediates neuroprotection via the inhibition of apoptosis (Figure 4) [110]. The upregulation of PDE5 expression in the brains of AD patients and the subsequent drop in cGMP levels have been linked to the elevation of Aβ amyloid peptide, whose deposition in the brain is the main hallmark of AD [111]. Sabayan et al. described PDE5-Is as disease-modifying agents against AD and proposed three main mechanisms for their action: (i) vasodilation, which improves or maintains cerebrovascular endothelial function preventing Aβ amyloid accumulation; (ii) cGMP-dependent rise in acetylcholine (ACh) levels in the cortex, striatum, and other areas of the brain, reversing low-ACh associated memory and cognitive deficits in AD, and finally (iii) inhibition of apoptosis and facilitation of neurogenesis averting neuronal loss (Figure 4) [112]. For example, chronic administration of sildenafil completely reversed cognitive impairment in Tg2576 transgenic mice without changing Aβ load. The underlying mechanism involved suppression of tau hyperphosphorylation and inhibition of glycogen synthase kinase 3β (GSK3β) and cyclin-dependent kinase 5 (CDK5) [113]. In addition, Puzzo et al. and Zhang et al. showed that chronic administration of sildenafil in amyloid precursor protein/presenilin-1 (APP/PS1) transgenic mice could reverse AD-related cognitive deficits and synaptic dysfunction via improving cGMP/PKG/CREB signaling, inhibiting neuroinflammation and reducing hippocampal Aβ levels [114][115][114,115]. Chronic treatment with tadalafil even exhibited a higher beneficial effect, probably due to its longer half-life and could improve spatial memory in the J20 mouse model of AD by decreasing tau protein via the activation of the AKT/GSK3β pathway [116]. Most recently, mirodenafil was reported to ameliorate Aβ-induced AD pathology and improve cognitive behavior in the APP-C105 mouse model through the modulation of the cGMP/PKG/CREB signaling pathway, GSK-3β activity, glucocorticoid receptor transcriptional activity and Wnt/β-catenin signaling in neuronal cells (Figure 4) [107]. Preclinical studies proved that PDE5-Is could boost memory and synaptic plasticity by augmenting the NO/cGMP/PKG pathway [107][117][107,117]. In mouse models with induced cognitive deficits, sildenafil could improve novel object recognition, ameliorate cognitive impairment and upregulate the brain-derived neurotrophic factor (BDNF), contributing to neuroprotective effects [118][119][118,119]. Another study showed the potential of sildenafil to defy neurological stress, increase neuroprotection and restore cognitive functions in the hippocampus region of noise alone-induced mice via modulation of cGMP/PKG/CREB and p25/CDK5 pathways and induction of various free radical scavengers in the brain of stressed mice [120]. A similar alleviation n of oxidative stress in the hippocampus of aged mice has been observed upon chronic tadalafil administration as well (Figure 4) [121]. Very recent reviews by Liu et al. [122] and Zuccarello et al. [123] summarized clinical trials of PDE5-Is in cognition and AD. However, none of the investigated drugs has reached the market for those indications so far. Numerous animal models investigated the potential role of PDE5 inhibition in stroke. In these studies, PDE5-Is could induce angiogenesis, enhance cerebral blood flow to the ischemic region, increase neurogenesis and advanced functional post-stroke recovery [124][125][126][124,125,126]. In particular, sildenafil treatment for two weeks (25 mg daily) was proven safe in patients who suffered mild to moderate strokes [127]. Additionally, tadalafil could attenuate ischemia-induced short-term memory impairment by suppressing ischemia-induced neuronal apoptosis [128]. Further mechanisms for PDE5 inhibition-induced neurogenesis have been reported and include AKT/GSK3β phosphorylation [129] or triggering proliferation of neural stem cells (NSC) via a mitogen-activated protein kinase (MAPK) dependent signaling cascade [130]. Moreover, preclinical studies have provided further evidence of sildenafil’s neuroprotective potential observed against Aβ amyloid-induced mitochondrial toxicity [131]. Additionally, 3-nitropropionic acid-induced behavioral and biochemical toxicities in a Huntington’s disease rat model [132]. Interestingly, a clinical study showed that single-dose sildenafil could improve regional cerebrovascular reactivity deficits in chronic traumatic brain injury patients as well [133]. Sildenafil has also been reported to promote efficient reconstitution of the myelin sheath and govern the inflammatory processes involved in demyelination models of MS [134]. Sildenafil could also normalize experimental autoimmune encephalomyelitis in MS mouse models [135]. Administration of sildenafil or tadalafil could yield significant anxiolytic-like effects in rodent genetic models of depression as well due to chronic activation of the NO/cGMP system [136][137][136,137]. Another reported mechanism for the antidepressant-like effect of sildenafil involved the activation of the oxytocin [138]. Jaumann et al. unveiled a potential protective role of activated cGMP/protein kinase cGMP-dependent 1/poly (ADP-ribose) polymerase (cGMP/PRKG1/PARP) signaling in response to traumas in cochlea sensory cells of various animal models. These data suggested PDE5 as a valid target for the improvement of NIHL. In particular, treatment of rodent models with vardenafil before or 6 h after acoustic trauma was shown to diminish auditory-evoked brain stream response thresholds in all frequency ranges tested [139]. Several animal studies have also proposed a beneficial pain-relieving effect of PDE5-Is in models of lesional [140][141][140,141] or metabolic neuropathic pain [142]. Sildenafil could ameliorate neuropathic pain symptoms in patients with diabetic peripheral neuropathy [143] and showed an antinociceptive effect in Sprague–Dawley male rats’ neuropathic pain models [144]. Mechanistically, this analgesic effect has been correlated to cGMP-dependent enhanced release of gamma-aminobutyric acid (GABA) [144].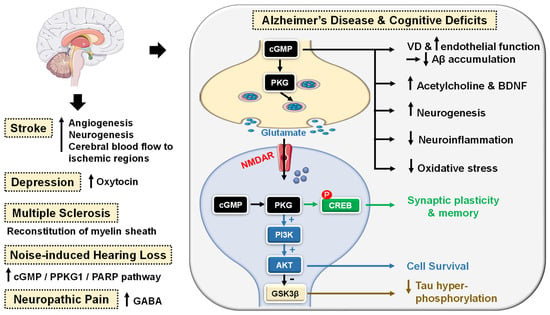 Figure 4. Emerging central nervous system (CNS)-related indications of PDE5 inhibitors. In Alzheimer’s disease (AD) and cognitive deficiency disease models, PDE5 inhibition increases presynaptic cGMP levels, which, through PKG activation, enhances the release of glutamate and activates N-methyl-D-aspartate receptors (NMDAR). On the other hand, postsynaptic PKG activates transcription factor cyclic adenosine monophosphate (cAMP) response element-binding element (CREB), promoting neurotransmission, synaptic plasticity and memory consolidation. PKG also activates the PI3K/AKT signaling pathway that mediates neuroprotection via the inhibition of apoptosis and also suppresses tau hyper-phosphorylation via inhibition of glycogen synthase kinase-3 beta (GSK3β). Elevated cGMP levels exhibit other cognitive enhancement mechanisms, such as vasodilation, which improves or maintains cerebrovascular endothelial function, preventing Aβ amyloid accumulation, rise in acetylcholine (ACh) and brain-derived neurotrophic factor (BDNF) levels in the cortex, striatum, and other areas of the brain, facilitation of neurogenesis, suppression of neuroinflammation and oxidative stress, all averting neuronal loss. In strokes, PDE5-Is could induce angiogenesis and neurogenesis and enhance cerebral blood flow to ischemic regions. PDE5-Is have anxiolytic effects in part due to enhanced oxytocin release. Moreover, PDE5-Is can promote efficient reconstitution of the myelin sheath and govern the Inflammatory processes involved in demyelination models of multiple sclerosis. PDE5-Is are also beneficial in noise-induced hearing loss via activating cGMP/protein kinase cGMP-dependent 1/poly (ADP-ribose) polymerase (cGMP/PRKG1/PARP) signaling in response to traumas in cochlea sensory cells. PDE5-Is exhibit pain-relieving effects in neuropathic pain models via enhanced release of gamma-aminobutyric acid (GABA). P = phosphorylation.
Figure 4. Emerging central nervous system (CNS)-related indications of PDE5 inhibitors. In Alzheimer’s disease (AD) and cognitive deficiency disease models, PDE5 inhibition increases presynaptic cGMP levels, which, through PKG activation, enhances the release of glutamate and activates N-methyl-D-aspartate receptors (NMDAR). On the other hand, postsynaptic PKG activates transcription factor cyclic adenosine monophosphate (cAMP) response element-binding element (CREB), promoting neurotransmission, synaptic plasticity and memory consolidation. PKG also activates the PI3K/AKT signaling pathway that mediates neuroprotection via the inhibition of apoptosis and also suppresses tau hyper-phosphorylation via inhibition of glycogen synthase kinase-3 beta (GSK3β). Elevated cGMP levels exhibit other cognitive enhancement mechanisms, such as vasodilation, which improves or maintains cerebrovascular endothelial function, preventing Aβ amyloid accumulation, rise in acetylcholine (ACh) and brain-derived neurotrophic factor (BDNF) levels in the cortex, striatum, and other areas of the brain, facilitation of neurogenesis, suppression of neuroinflammation and oxidative stress, all averting neuronal loss. In strokes, PDE5-Is could induce angiogenesis and neurogenesis and enhance cerebral blood flow to ischemic regions. PDE5-Is have anxiolytic effects in part due to enhanced oxytocin release. Moreover, PDE5-Is can promote efficient reconstitution of the myelin sheath and govern the Inflammatory processes involved in demyelination models of multiple sclerosis. PDE5-Is are also beneficial in noise-induced hearing loss via activating cGMP/protein kinase cGMP-dependent 1/poly (ADP-ribose) polymerase (cGMP/PRKG1/PARP) signaling in response to traumas in cochlea sensory cells. PDE5-Is exhibit pain-relieving effects in neuropathic pain models via enhanced release of gamma-aminobutyric acid (GABA). P = phosphorylation.5.2.3. Cardiovascular Diseases
Cardiomyocytes normally express a minimal basal level of PDE5. However, cardiac PDE5 expression was reported to be upregulated in hypertrophic, dilated, and ischemic cardiomyopathy and in congestive heart failure [47][145][146][47,145,146]. The protective effects of PDE5-Is against myocardial infarction (MI), cardiac ischemic and reperfusion (I/R) injury were validated in many in vitro studies with sildenafil [147], tadalafil [148][149][148,149], and vardenafil [150]. When given either prior to occlusion or at reperfusion, these PDE5-Is could reduce infarct size, attenuate cardiac hypertrophy, improve left ventricular (LV) function and prevent progression to heart failure. In a mouse model, sildenafil exhibited a preconditioning effect to protect the heart against necrosis and apoptosis [151]. Another study suggested that the cardioprotective effect of sildenafil in female mice is estrogen-dependent as ovariectomy suppressed its anti-hypertrophic effect [152]. Intramyocardial transplantation of human adipose stem cells (ASCs) is regarded as a potential treatment for post-ischemic heart failure. Hoke et al. showed that preconditioning of ASCs with sildenafil could trigger the release of significantly high levels of pro-angiogenic or pro-survival growth factors, which enhance ASCs survival and therapeutic efficacy in cardiac ischemic microenvironment, allowing successful cardiac regeneration [153]. Tadalafil also showed cardioprotective effects via PKG-dependent generation of hydrogen sulfide [154]. Moreover, tadalafil was suggested to be clinically beneficial in metabolic syndrome (MetS) patients who are at high risk for CVS diseases where it improved insulin sensitivity, lowered circulating lipids, improved LV diastolic dysfunction and protected against I/R injury in MetS mice [155]. PDE5-Is manifested more significant protective effects against advanced heart failure (HF) with reduced ejection fraction than in HF with preserved ejection fraction [156]. Sildenafil could suppress chamber and myocyte hypertrophy and reverse preestablished hypertrophy in mice exposed to chronic pressure overload. This anti-hypertrophic effect was mediated by the deactivation of multiple signaling pathways, including the calcineurin/nuclear factor of activated T-cells (NFAT), PI3K/AKT, and ERK1/2 signaling pathways [157]. Furthermore, several clinical studies have confirmed the potential role of sildenafil in improving cardiac output, endothelial function, muscle perfusion, and exercise ventilatory and aerobic efficiencies in systolic HF patients [158][159][160][158,159,160]. Moreover, prophylactic treatment with either sildenafil or tadalafil improved cardiac contractile function and survival by attenuating doxorubicin-induced apoptosis and cardiac oxidative stress without interfering with the antitumor efficacy of doxorubicin in both in vitro and in vivo tumor models [161][162][161,162]. PDE5 inhibition could govern two crucial vascular manifestations of essential hypertension as well via diminishing blood pressure and improving arterial stiffness and endothelial dysfunction [163]. In addition, sildenafil elicited a significant decrease in inducible ventricular tachycardia and ventricular fibrillation in animal models and demonstrated protection against ventricular arrhythmias associated with the early stages of cardiac ischemia or following MI [164][165][164,165]. PDE5-Is could also inhibit platelet activation and aggregation [166][167][166,167]. Sildenafil, in particular, was demonstrated to (i) improve coronary patency in an animal model [168], (ii) reduce thrombosis, thromboembolic events, and the risk of thrombotic strokes in a clinical study [169], and (iii) potentiate the anti-aggregation effect of NO donors via cGMP-dependent and independent pathways [170]. Owing to their vasoactive effects, both sildenafil and tadalafil showed advantages in minimizing skin flap necrosis and in preventing extremity and flap ischemia in patients with Raynaud’s phenomenon and with scleroderma [171][172][171,172]. Kloner et al. thoroughly investigated the cardiovascular safety profile of PDE5-Is published in the last two decades and confirmed their safety [173]. Cardio protection achieved by PDE5-Is is mainly attributed to restoring high cGMP levels in cardiomyocytes that govern diverse cardioprotective mechanisms as follows (Figure 5): (i) vascular tone regulation and release of endogenous cardioprotective molecules, such as adenosine, bradykinin and phenylephrine from endothelial cells [174], (ii) PKG-dependent opening of mitochondrial and sarcolemmal ATP-sensitive potassium channels modulating calcium homeostasis and survival of cardiomyocytes, preventing post-infarct LV remodeling and reducing infarct size [175][176][175,176], (iii) PKG-dependent suppression of adrenergic drive which reduces nerve growth factor leading to anti-arrhythmic effects [164], (iv) ischemic post-conditioning protection against MI via PKG-dependent enhancement of Na+/K+-ATPase activity [177] and inhibition of Na+/H+-exchanger, delaying normalization of pH during reperfusion [178], (v) suppression of protein kinase C (PKC) and calcineurin culminating in improved contractility and protection against HF [179], (vi) improving mitochondrial ultrastructure and function via increased sirtuin-3 (Sirt3) protein expression and decreased peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α) acetylation protecting against post-infarction HF [180], and (vii) inhibition of RhoA/Rho-kinase pathway [181].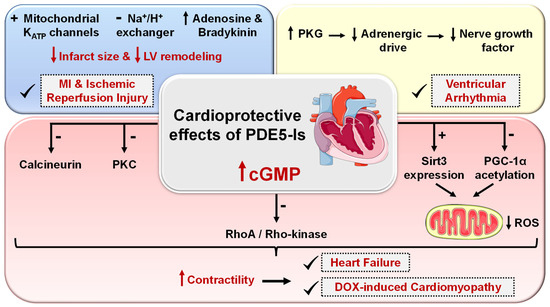 Figure 5. Cardioprotective effects of PDE5 inhibitors. PDE5-Is restore high cGMP levels in cardiomyocytes that govern diverse downstream cardioprotective mechanisms: (i) PKG-dependent opening of mitochondrial and sarcolemmal ATP-sensitive potassium channels, inhibition of Na+/H+-exchanger and release of endogenous cardioprotective molecules, such as adenosine, bradykinin from endothelial cells; resulting in reduced infarct size and hampered post-infarct left ventricular (LV) remodeling. All are beneficial for ischemic post-conditioning protection against myocardial infarction (MI) and ischemic reperfusion (I/R) injury, (ii) PKG-dependent suppression of adrenergic drive which reduces nerve growth factor leading to anti-arrhythmic effects, (iii) suppression of protein kinase C (PKC), calcineurin and RhoA/Rho-kinase pathways and (vi) suppression of oxidative stress and improving mitochondrial ultrastructure and function via increased sirtuin-3 (Sirt3) protein expression and decreased peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α) acetylation, all culminating in improved cardiac contractility and protection against heart failure (HF) and doxorubicin(dox)-induced cardiomyopathy.
Figure 5. Cardioprotective effects of PDE5 inhibitors. PDE5-Is restore high cGMP levels in cardiomyocytes that govern diverse downstream cardioprotective mechanisms: (i) PKG-dependent opening of mitochondrial and sarcolemmal ATP-sensitive potassium channels, inhibition of Na+/H+-exchanger and release of endogenous cardioprotective molecules, such as adenosine, bradykinin from endothelial cells; resulting in reduced infarct size and hampered post-infarct left ventricular (LV) remodeling. All are beneficial for ischemic post-conditioning protection against myocardial infarction (MI) and ischemic reperfusion (I/R) injury, (ii) PKG-dependent suppression of adrenergic drive which reduces nerve growth factor leading to anti-arrhythmic effects, (iii) suppression of protein kinase C (PKC), calcineurin and RhoA/Rho-kinase pathways and (vi) suppression of oxidative stress and improving mitochondrial ultrastructure and function via increased sirtuin-3 (Sirt3) protein expression and decreased peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α) acetylation, all culminating in improved cardiac contractility and protection against heart failure (HF) and doxorubicin(dox)-induced cardiomyopathy.5.2.4. Kidney Diseases
Coskuner and coauthor [17] and Afsar et al. [182] thoroughly investigated the renoprotective benefits of PDE5-Is in kidney-related clinical conditions, such as diabetic or nephrotoxic nephropathy, renal ischemia/reperfusion injury, renovascular hypertension and chronic kidney disease. Most reported preclinical studies highlighted a promising potential of PDE5-Is to improve renal function and histopathological changes via collaborative mechanisms, including antioxidative, anti-inflammatory, anti-apoptotic, antifibrotic pathways along with suppression of DNA damage and improving renal blood flow, NOS levels, endothelial function and mitochondrial biogenesis. Most recently, tadalafil was also reported to avert the onset of ureter inflammation and urothelial degeneration in a unilateral ureteral obstruction animal model via modulation of various histopathologic and biochemical changes [183].5.2.5. Cystic Fibrosis
Cystic fibrosis (CF) is a disease that is caused by a mutation in the CF transmembrane conductance regulator (CFTR) gene “F508del allele” that encodes the main chloride channel expressed in epithelia, which leads to a reduced transepithelial chloride transport in multiple organs, such as pancreas, intestine, kidney, liver and most significantly lungs. This results in abnormal mucociliary clearance and endosomal hyper-acidification along with obstruction, infection and excessive proinflammatory responses that progressively damage the respective organ function and structure [184]. Several preclinical and clinical studies highlighted that PDE5-Is can correct the majority of the known pathological defects in CF, where tadalafil showed the highest efficacy, while vardenafil granted prolonged effects after a single therapeutic dose [185][186][185,186]. The efficacy of PDE5-Is in CF could be correlated to one or more of the following mechanisms: (i) correction of the mislocalization of the mutant CFTR protein, restoring normal transepithelial chloride transport [187][188][189][187,188,189], (ii) normalizing the excessive proinflammatory responses via downregulation of M1 markers, tumor necrosis factor (TNF)-α and inducible NOS-2 [190][191][190,191], (iii) reversing endosomal hyper-acidification via elevating cGMP levels [192], (iv) improving endothelial function via promoting NOS-3 phosphorylation in endothelial cells [193], and (v) reducing adhesion of bacterial pathogens to respiratory epithelial cells [190].5.2.6. Diabetes
Das et al. have summarized the potential protective roles of PDE5-Is against several diabetes-related pathologies including (i) prevention of diabetic neuropathy and vasculopathy via improving endothelial function, (ii) protection against I/R injury in diabetic heart via an AMP-activated protein kinase/Sirt1/PGC-1α (AMPK/Sirt1/PGC-1α) cytoprotective signaling cascade, along with (iii) antioxidant and anti-inflammatory effects in diabetic hearts [86]. A meta-analysis of randomized controlled trials has also validated PDE5-Is as effective and safe medications for the treatment of sexual dysfunction in patients with diabetes mellitus suffering from ED [194]. Most recently, a combination of tadalafil and hydrochloroquine successfully improved several Type 2 diabetes-related clinical parameters, including a drop in fasting blood glucose and lipid levels, a rise in plasma insulin and insulin-like growth factor-1 levels and improved insulin sensitivity. Interestingly, pretreatment with the same combination showed a potential to diminish the rate and severity of COVID-19 infection in vulnerable diabetic patients [195].5.2.7. Miscellaneous Indications
Several studies have demonstrated the efficacy of the combined administration of sildenafil with selective serotonin reuptake inhibitors (SSRIs), such as paroxetine and sertraline, for the treatment of premature ejaculation [196]. Moreover, PDE5-Is prompted penile rigidity and recovery of erections in the post-ejaculatory period [197]. Details of related preclinical and clinical trials were further elaborated by the reviews [23][198][23,198]. Long-term chronic administration of PDE5-Is could also avert the progression of fibrotic plaques and halt corporal fibrosis in animal models of Peyronie’s disease [199][200][199,200]. In addition, prolonged administration of low-dose PDE5-Is exhibited a promising beneficial effect in the treatment of male infertility. Sildenafil and vardenafil, in particular, could enhance Leydig cells’ secretory and steroidogenic functions, augmenting sperm concentration and the percentages of motile and morphologically normal sperm [201][202][203][201,202,203]. An increase in serum testosterone levels by both inhibitors has been reported as well [204]. Interestingly, tadalafil was proven safe to improve selective fetal growth restriction, a condition of twin pregnancy in which the development of one fetus is restricted, without severe side effects in the mothers or neonates [205]. Most recently, Isidori et al. collaborated evidence possibly linking the NO/cGMP/PDE5 axis to the pathophysiology of coronavirus disease (COVID-19) and suggested the repurposing of PDE5-Is as a treatment strategy to halt the progression of COVID-19 via diverse immunomodulatory mechanisms [206]. All reported FDA-approved and emerging uses of PDE5-Is are summarized in Figure 6.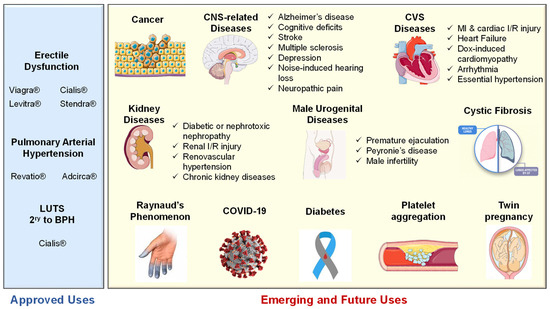 Figure 6.Summary of approved and emerging/future uses of PDE5 inhibitors.
Figure 6.Summary of approved and emerging/future uses of PDE5 inhibitors.5.3. Side Effects and Contraindications of PDE5 Inhibitors
The use of PDE5-Is is usually associated with some common side effects, which include headache, flushing, dyspepsia, visual disturbances, back pain, myalgia, tachycardia, and nasal congestion [207]. Most of these side effects are due to the inhibition of PDEs other than PDE5, visual disturbances are associated with PDE6 inhibition and back pain and myalgia are attributed to the inhibition of PDE11. Nevertheless, these side effects rarely led to discontinuation of the treatment. Other less known, seldom encountered serious side effects have been reported concomitant to the use of PDE5-Is are highlighted in the following lines.- (i)
-
Although PDE5 is reported as a promising target for anti-cancer therapy, as explained earlier, the prolonged use of PDE5-Is has been linked to an increased risk of melanoma. Lie and co-workers reported an association between sildenafil use and an increased risk of melanoma in a prospective cohort study conducted on 25,848 men [208]. Several other cohorts and case-control studies have also reported a correlation between the use of sildenafil and tadalafil and the increased risk of melanoma [209][210][209,210]. However, this association between the prolonged use of PDE5-Is and the development of cancer was only reported for melanoma; even the risk of other types of skin cancer, such as squamous cell carcinoma and basal cell carcinoma, was not correlated to the use of PDE5-Is [211].
- (ii)
-
Visual disturbances have been usually reported with the use of PDE5-Is because of PDE6 inhibition. However, several studies have reported more serious ophthalmologic side effects associated with the use of PDE5-Is, which include non-arteritic anterior ischemic optic neuropathy (NAION), which may eventually lead to vision loss [212]. Two case-crossover studies have shown a two-fold increase in the risk of NAION in men using PDE5-Is, and currently, all PDE5Is (Viagra®, Cialis®, Levitra® and Spedra®) mention NAION as a caution in their summary of product characteristics [213][214][213,214].
- (iii)
-
Moreover, sensorineural hearing loss (SSHL) has been associated with the prolonged use of PDE5-Is. Two in vivo studies have shown that the prolonged use of sildenafil could lead to hearing loss in mice and rats [215][216][215,216]; in addition, published trials and pharmacovigilance agencies reported 47 cases of SSHL as a result of prolonged administration of sildenafil [217], and more specifically, Maddox et al. reported two cases of SSHL due to daily use of tadalafil 10 mg and sildenafil 50 mg + tadalafil 10 mg use where both patients did not recover after a follow-up [218]. Both NAION and SSHL are of unknown pathophysiology.
- (iv)
-
Priapism (prolonged erection of the penis) is another less common side effect reported with the prolonged use of PDE5-Is, as only a few cases have been reported for priapism associated with the use of PDE5-Is [219]. The risk of priapism increases in the case of concomitant use of other ED medications along with the PDE5-Is.