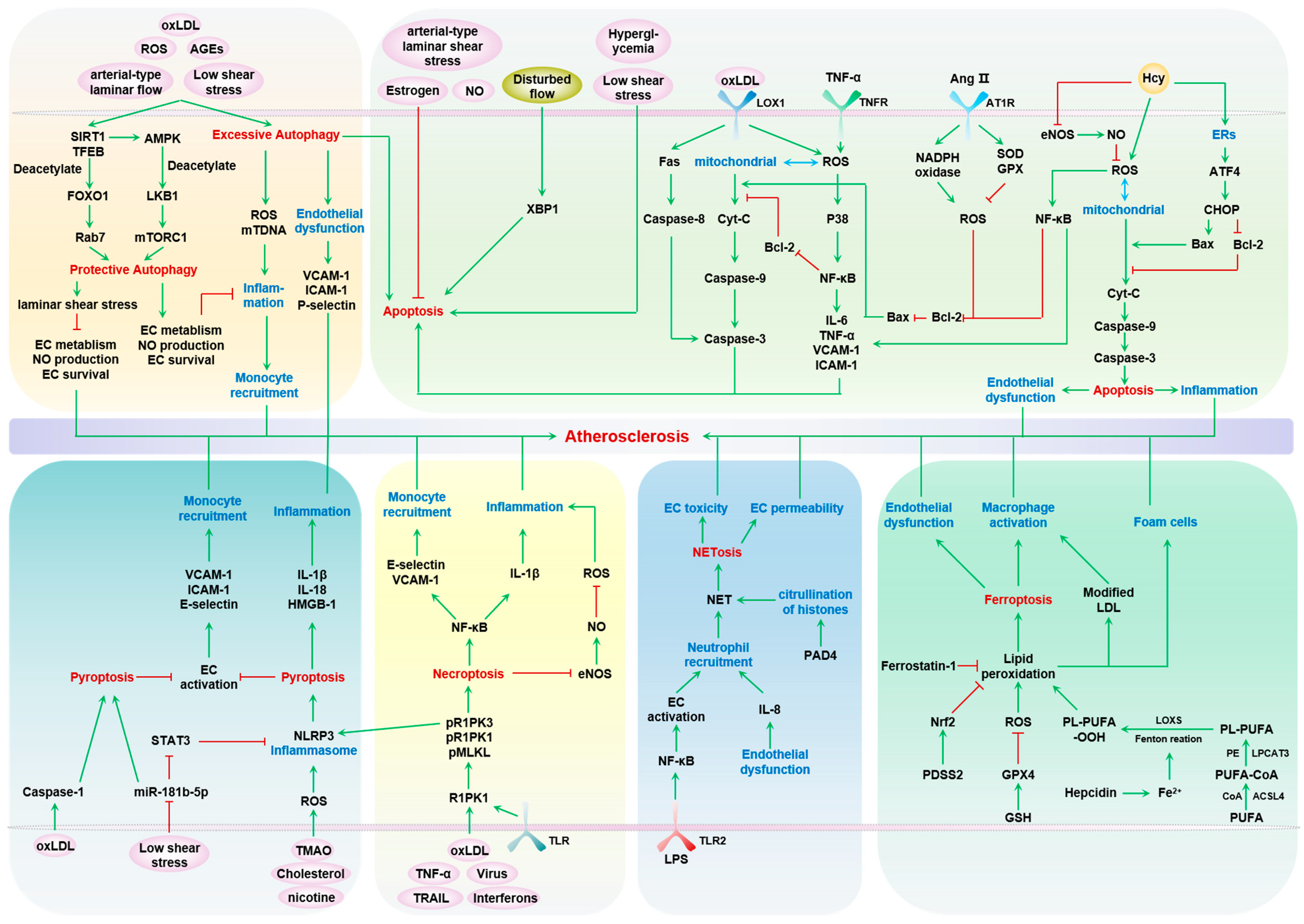
Endothelial cells (ECs) form the inner linings of blood vessels, and are directly exposed to endogenous hazard signals and metabolites in the circulatory system. The senescence and death of ECs are not only adverse outcomes, but also causal contributors to endothelial dysfunction, an early risk marker of atherosclerosis. The pathophysiological process of EC senescence involves both structural and functional changes and has been linked to various factors, including oxidative stress, dysregulated cell cycle, hyperuricemia, vascular inflammation, and aberrant metabolite sensing and signaling. Multiple forms of EC death have been documented in atherosclerosis, including autophagic cell death, apoptosis, pyroptosis, NETosis, necroptosis, and ferroptosis. Despite this, the molecular mechanisms underlying EC senescence or death in atherogenesis are not fully understood.
- atherosclerosis
- endothelial cell
- endothelial cell senescence
- endothelial cell death
1. Endothelial Cell Death and Atherosclerosis
| Cell Death | Morphological Features | Biochemical Features | Key Biomarkers | Hallmarks | Related Signaling Molecules and Pathways |
|
|---|---|---|---|---|---|---|
| Autophagy | Formation of double-membraned autolysosomes |
Increased lysosomal activity |
ATG5, ATG7, DRAM3, TFEB |
LC3-II/I, autophagic vesicles, autophagic flow |
PI3K-AKT-mTOR, MAPK-ERK1/2 pathway |
[4][5] |
| Apoptosis | Cell membrane blebbing, cell shrinkage, nuclear fragmentation, chromatin condensation, DNA fragmentation, and apoptotic body formation |
Activation of caspases, oligonucleosomal DNA fragmentation, modification of nuclear polypeptides |
Caspase, p53, Fas, Bcl-2, Bax |
Cell morphology, annexin V/PI, mitochondrial membrane potential, cytochrome C |
Death receptor, mitochondrial, endoplasmic reticulum pathway, caspase, p53, Bcl-2-mediated pathway |
[6][7] |
| Anoikis | Insufficient cell–matrix interactions | Integrins interact with ECM components to form adhesion complexes |
Bcl-2, Bcl-XL, Mcl1, Bax, Bak, Bok, Bid, Bik, Bmf, Noxa, Bad, Bim, Puma |
Caspases activation, DNA fragmentation | JNK, ERK, PI3K, PKB/AKT pathway, integrin, caveolin-1 |
[8][9] |
| Pyroptosis | Apoptosis-like chromatin condensation and DNA fragmentation in the early stage, karyopyknosis, necrosis-like cell membrane pore formation, cellular swelling, membrane rupture |
Dependent on caspase-1 and proinflammatory cytokine releases |
Caspase-1, IL-1β, IL-18 |
Gasdermin D, caspase-4, cell morphology |
Caspase-1, NLRP3-mediated pathway |
[10][11] |
| NETosis | Enzymes from granules translocate to the nucleus, chromatin de-condensation, internal membranes break down, cytolysis releases NETs |
NETs generation | LPS, GM-CSF, IL8, PAD4, HMGB1, TF, MMP9, PMA, TGFβ, MPO, NE, RAGE, Histone, Anti-DsDNA |
Promoting the transcription and translation of NE, MPO, and PAD4 |
Raf-MEK-ERK, TLR2-fibronectin, caspase-4/5-GSDMD pathway |
[12][13] |
| Necroptosis | Rupture of the cellular membrane, progressively translucent cytoplasm and swelling of organelles, moderate chromatin condensation |
Drop in ATP levels, Activation of RIP1, RIP3, MLKL |
LEF1, RIP1, RIP3 | RIPK1, RIPK3 | TNFα, TNFR1, TLR3, TRAIL, FasL, ROS, PKC-MAPK-AP1-mediated pathway |
[14][15] |
| Ferroptosis | Small mitochondria with condensed mitochondrial membrane densities, reduction in or vanishing of mitochondria crista, outer mitochondrial membrane rupture |
Inhibition of xCT and reduced GSH, inhibition of GPX4 Iron accumulation, lipid peroxidation |
GPX4, Nrf2, LSH, TFR1, xCT |
Fe2+, ROS, GSH, lipid peroxidation |
MVA, HSF1-HSPB1, p62-Keap1-Nrf2 pathway; LSH pathway |
[16][17] |
2. Autophagy—Protective and Detrimental
3. Apoptosis
4. Necroptosis
5. Pyroptosis
6. Ferroptosis
7. NETosis
8. Complexity and Limitations of Targeting Endothelial Cell Death in Atherosclerosis
References
- Gautier, E.L.; Huby, T.; Witztum, J.L.; Ouzilleau, B.; Miller, E.R.; Saint-Charles, F.; Aucouturier, P.; Chapman, M.J.; Lesnik, P. Macrophage apoptosis exerts divergent effects on atherogenesis as a function of lesion stage. Circulation 2009, 119, 1795–1804.
- Proudfoot, D.; Skepper, J.N.; Hegyi, L.; Bennett, M.R.; Shanahan, C.M.; Weissberg, P.L. Apoptosis regulates human vascular calcification in vitro: Evidence for initiation of vascular calcification by apoptotic bodies. Circ. Res. 2000, 87, 1055–1062.
- Li, M.; Wang, Z.W.; Fang, L.J.; Cheng, S.Q.; Wang, X.; Liu, N.F. Programmed cell death in atherosclerosis and vascular calcification. Cell Death Dis. 2022, 13, 467.
- Dikic, I.; Elazar, Z. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 349–364.
- Zhao, F.; Satyanarayana, G.; Zhang, Z.; Zhao, J.; Ma, X.L.; Wang, Y. Endothelial Autophagy in Coronary Microvascular Dysfunction and Cardiovascular Disease. Cells 2022, 11, 2081.
- Choy, J.C.; Granville, D.J.; Hunt, D.W.; McManus, B.M. Endothelial cell apoptosis: Biochemical characteristics and potential implications for atherosclerosis. J. Mol. Cell. Cardiol. 2001, 33, 1673–1690.
- Nagata, S. Apoptosis and Clearance of Apoptotic Cells. Annu. Rev. Immunol. 2018, 36, 489–517.
- Michel, J.B. Anoikis in the cardiovascular system: Known and unknown extracellular mediators. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 2146–2154.
- Taddei, M.L.; Giannoni, E.; Fiaschi, T.; Chiarugi, P. Anoikis: An emerging hallmark in health and diseases. J. Pathol. 2012, 226, 380–393.
- Shi, J.; Gao, W.; Shao, F. Pyroptosis: Gasdermin-Mediated Programmed Necrotic Cell Death. Trends Biochem. Sci. 2017, 42, 245–254.
- Ju, J.; Liu, Y.; Liang, H.; Yang, B. The role of pyroptosis in endothelial dysfunction induced by diseases. Front. Immunol. 2022, 13, 1093985.
- Zdanyte, M.; Borst, O.; Münzer, P. NET-(works) in arterial and venous thrombo-occlusive diseases. Front. Cardiovasc. Med. 2023, 10, 1155512.
- Thiam, H.R.; Wong, S.L.; Wagner, D.D.; Waterman, C.M. Cellular Mechanisms of NETosis. Annu. Rev. Cell Dev. Biol. 2020, 36, 191–218.
- Weinlich, R.; Oberst, A.; Beere, H.M.; Green, D.R. Necroptosis in development, inflammation and disease. Nat. Rev. Mol. Cell Biol. 2017, 18, 127–136.
- Zhang, X.; Ren, Z.; Xu, W.; Jiang, Z. Necroptosis in atherosclerosis. Clin. Chim. Acta Int. J. Clin. Chem. 2022, 534, 22–28.
- Zhang, H.; Zhou, S.; Sun, M.; Hua, M.; Liu, Z.; Mu, G.; Wang, Z.; Xiang, Q.; Cui, Y. Ferroptosis of Endothelial Cells in Vascular Diseases. Nutrients 2022, 14, 4506.
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 266–282.
- Aman, Y.; Schmauck-Medina, T.; Hansen, M.; Morimoto, R.I.; Simon, A.K.; Bjedov, I.; Palikaras, K.; Simonsen, A.; Johansen, T.; Tavernarakis, N.; et al. Autophagy in healthy aging and disease. Nat. Aging 2021, 1, 634–650.
- Pyo, J.O.; Yoo, S.M.; Ahn, H.H.; Nah, J.; Hong, S.H.; Kam, T.I.; Jung, S.; Jung, Y.K. Overexpression of Atg5 in mice activates autophagy and extends lifespan. Nat. Commun. 2013, 4, 2300.
- LaRocca, T.J.; Henson, G.D.; Thorburn, A.; Sindler, A.L.; Pierce, G.L.; Seals, D.R. Translational evidence that impaired autophagy contributes to arterial ageing. J. Physiol. 2012, 590, 3305–3316.
- Lesniewski, L.A.; Seals, D.R.; Walker, A.E.; Henson, G.D.; Blimline, M.W.; Trott, D.W.; Bosshardt, G.C.; LaRocca, T.J.; Lawson, B.R.; Zigler, M.C.; et al. Dietary rapamycin supplementation reverses age-related vascular dysfunction and oxidative stress, while modulating nutrient-sensing, cell cycle, and senescence pathways. Aging Cell 2017, 16, 17–26.
- LaRocca, T.J.; Gioscia-Ryan, R.A.; Hearon, C.M., Jr.; Seals, D.R. The autophagy enhancer spermidine reverses arterial aging. Mech. Ageing Dev. 2013, 134, 314–320.
- Wang, J.; Wang, W.N.; Xu, S.B.; Wu, H.; Dai, B.; Jian, D.D.; Yang, M.; Wu, Y.T.; Feng, Q.; Zhu, J.H.; et al. MicroRNA-214-3p: A link between autophagy and endothelial cell dysfunction in atherosclerosis. Acta Physiol. 2018, 222, e12973.
- Chen, Y.; Zeng, A.; He, S.; He, S.; Li, C.; Mei, W.; Lu, Q. Autophagy-Related Genes in Atherosclerosis. J. Healthc. Eng. 2021, 2021, 6402206.
- Patella, F.; Neilson, L.J.; Athineos, D.; Erami, Z.; Anderson, K.I.; Blyth, K.; Ryan, K.M.; Zanivan, S. In-Depth Proteomics Identifies a Role for Autophagy in Controlling Reactive Oxygen Species Mediated Endothelial Permeability. J. Proteome Res. 2016, 15, 2187–2197.
- Zhu, L.; Wu, G.; Yang, X.; Jia, X.; Li, J.; Bai, X.; Li, W.; Zhao, Y.; Li, Y.; Cheng, W.; et al. Low density lipoprotein mimics insulin action on autophagy and glucose uptake in endothelial cells. Sci. Rep. 2019, 9, 3020.
- Yuan, P.; Hu, Q.; He, X.; Long, Y.; Song, X.; Wu, F.; He, Y.; Zhou, X. Laminar flow inhibits the Hippo/YAP pathway via autophagy and SIRT1-mediated deacetylation against atherosclerosis. Cell Death Dis. 2020, 11, 141.
- Torisu, K.; Singh, K.K.; Torisu, T.; Lovren, F.; Liu, J.; Pan, Y.; Quan, A.; Ramadan, A.; Al-Omran, M.; Pankova, N.; et al. Intact endothelial autophagy is required to maintain vascular lipid homeostasis. Aging Cell 2016, 15, 187–191.
- Xie, Y.; You, S.J.; Zhang, Y.L.; Han, Q.; Cao, Y.J.; Xu, X.S.; Yang, Y.P.; Li, J.; Liu, C.F. Protective role of autophagy in AGE-induced early injury of human vascular endothelial cells. Mol. Med. Rep. 2011, 4, 459–464.
- Zhang, X.; Yu, L.; Xu, H. Lysosome calcium in ROS regulation of autophagy. Autophagy 2016, 12, 1954–1955.
- Cho, K.; Choi, S.H. ASK1 Mediates Apoptosis and Autophagy during oxLDL-CD36 Signaling in Senescent Endothelial Cells. Oxidative Med. Cell. Longev. 2019, 2019, 2840437.
- Meng, Q.; Pu, L.; Lu, Q.; Wang, B.; Li, S.; Liu, B.; Li, F. Morin hydrate inhibits atherosclerosis and LPS-induced endothelial cells inflammatory responses by modulating the NFκB signaling-mediated autophagy. Int. Immunopharmacol. 2021, 100, 108096.
- Reglero-Real, N.; Pérez-Gutiérrez, L.; Yoshimura, A.; Rolas, L.; Garrido-Mesa, J.; Barkaway, A.; Pickworth, C.; Saleeb, R.S.; Gonzalez-Nuñez, M.; Austin-Williams, S.N.; et al. Autophagy modulates endothelial junctions to restrain neutrophil diapedesis during inflammation. Immunity 2021, 54, 1989–2004.e9.
- Vion, A.C.; Kheloufi, M.; Hammoutene, A.; Poisson, J.; Lasselin, J.; Devue, C.; Pic, I.; Dupont, N.; Busse, J.; Stark, K.; et al. Autophagy is required for endothelial cell alignment and atheroprotection under physiological blood flow. Proc. Natl. Acad. Sci. USA 2017, 114, E8675–E8684.
- Lu, H.; Fan, Y.; Qiao, C.; Liang, W.; Hu, W.; Zhu, T.; Zhang, J.; Chen, Y.E. TFEB inhibits endothelial cell inflammation and reduces atherosclerosis. Sci. Signal. 2017, 10, eaah4214.
- Kim, J.Y.; Mondaca-Ruff, D.; Singh, S.; Wang, Y. SIRT1 and Autophagy: Implications in Endocrine Disorders. Front. Endocrinol. 2022, 13, 930919.
- Xu, Y.; Wan, W. Acetylation in the regulation of autophagy. Autophagy 2023, 19, 379–387.
- Gu, S.; Zhou, C.; Pei, J.; Wu, Y.; Wan, S.; Zhao, X.; Hu, J.; Hua, X. Esomeprazole inhibits hypoxia/endothelial dysfunction-induced autophagy in preeclampsia. Cell Tissue Res. 2022, 388, 181–194.
- Dossou, A.S.; Basu, A. The Emerging Roles of mTORC1 in Macromanaging Autophagy. Cancers 2019, 11, 1422.
- Kim, Y.C.; Guan, K.L. mTOR: A pharmacologic target for autophagy regulation. J. Clin. Investig. 2015, 125, 25–32.
- Lee, I.H.; Cao, L.; Mostoslavsky, R.; Lombard, D.B.; Liu, J.; Bruns, N.E.; Tsokos, M.; Alt, F.W.; Finkel, T. A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc. Natl. Acad. Sci. USA 2008, 105, 3374–3379.
- Jiang, Q.; Hao, R.; Wang, W.; Gao, H.; Wang, C. SIRT1/Atg5/autophagy are involved in the antiatherosclerosis effects of ursolic acid. Mol. Cell. Biochem. 2016, 420, 171–184.
- Liu, J.; Bi, X.; Chen, T.; Zhang, Q.; Wang, S.X.; Chiu, J.J.; Liu, G.S.; Zhang, Y.; Bu, P.; Jiang, F. Shear stress regulates endothelial cell autophagy via redox regulation and Sirt1 expression. Cell Death Dis. 2015, 6, e1827.
- Zhu, L.; Duan, W.; Wu, G.; Zhang, D.; Wang, L.; Chen, D.; Chen, Z.; Yang, B. Protective effect of hydrogen sulfide on endothelial cells through Sirt1-FoxO1-mediated autophagy. Ann. Transl. Med. 2020, 8, 1586.
- Li, Y.; Jiang, X.; Zhang, Z.; Liu, J.; Wu, C.; Chen, Y.; Zhou, J.; Zhang, J.; Zhang, X. Autophagy promotes directed migration of HUVEC in response to electric fields through the ROS/SIRT1/FOXO1 pathway. Free Radic. Biol. Med. 2022, 192, 213–223.
- Xu, C.; Wang, L.; Fozouni, P.; Evjen, G.; Chandra, V.; Jiang, J.; Lu, C.; Nicastri, M.; Bretz, C.; Winkler, J.D.; et al. SIRT1 is downregulated by autophagy in senescence and ageing. Nat. Cell Biol. 2020, 22, 1170–1179.
- Hua, Y.; Zhang, J.; Liu, Q.; Su, J.; Zhao, Y.; Zheng, G.; Yang, Z.; Zhuo, D.; Ma, C.; Fan, G. The Induction of Endothelial Autophagy and Its Role in the Development of Atherosclerosis. Front. Cardiovasc. Med. 2022, 9, 831847.
- De Meyer, G.R.; Grootaert, M.O.; Michiels, C.F.; Kurdi, A.; Schrijvers, D.M.; Martinet, W. Autophagy in vascular disease. Circ. Res. 2015, 116, 468–479.
- Liu, Y.; Shoji-Kawata, S.; Sumpter, R.M., Jr.; Wei, Y.; Ginet, V.; Zhang, L.; Posner, B.; Tran, K.A.; Green, D.R.; Xavier, R.J.; et al. Autosis is a Na+,K+-ATPase-regulated form of cell death triggered by autophagy-inducing peptides, starvation, and hypoxia-ischemia. Proc. Natl. Acad. Sci. USA 2013, 110, 20364–20371.
- Chau, Y.P.; Lin, S.Y.; Chen, J.H.; Tai, M.H. Endostatin induces autophagic cell death in EAhy926 human endothelial cells. Histol. Histopathol. 2003, 18, 715–726.
- Kato, Y.; Furusyo, N.; Tanaka, Y.; Ueyama, T.; Yamasaki, S.; Murata, M.; Hayashi, J. The Relation between Serum Endostatin Level and Carotid Atherosclerosis in Healthy Residents of Japan: Results from the Kyushu and Okinawa Population Study (KOPS). J. Atheroscler. Thromb. 2017, 24, 1023–1030.
- Ärnlöv, J.; Ruge, T.; Ingelsson, E.; Larsson, A.; Sundström, J.; Lind, L. Serum endostatin and risk of mortality in the elderly: Findings from 2 community-based cohorts. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2689–2695.
- Zhang, C.; Qian, S.; Zhang, R.; Guo, D.; Wang, A.; Peng, Y.; Peng, H.; Li, Q.; Ju, Z.; Geng, D.; et al. Endostatin as a novel prognostic biomarker in acute ischemic stroke. Atherosclerosis 2020, 293, 42–48.
- Moulton, K.S.; Heller, E.; Konerding, M.A.; Flynn, E.; Palinski, W.; Folkman, J. Angiogenesis inhibitors endostatin or TNP-470 reduce intimal neovascularization and plaque growth in apolipoprotein E-deficient mice. Circulation 1999, 99, 1726–1732.
- Zeng, X.; Chen, J.; Miller, Y.I.; Javaherian, K.; Moulton, K.S. Endostatin binds biglycan and LDL and interferes with LDL retention to the subendothelial matrix during atherosclerosis. J. Lipid Res. 2005, 46, 1849–1859.
- Zeng, M.; Wei, X.; Wu, Z.; Li, W.; Zheng, Y.; Li, B.; Meng, X.; Fu, X.; Fei, Y. Simulated ischemia/reperfusion-induced p65-Beclin 1-dependent autophagic cell death in human umbilical vein endothelial cells. Sci. Rep. 2016, 6, 37448.
- Lu, Y.; Wang, Z.; Han, W.; Li, H. Zoledronate induces autophagic cell death in human umbilical vein endothelial cells via Beclin-1 dependent pathway activation. Mol. Med. Rep. 2016, 14, 4747–4754.
- Csordas, A.; Kreutmayer, S.; Ploner, C.; Braun, P.R.; Karlas, A.; Backovic, A.; Wick, G.; Bernhard, D. Cigarette smoke extract induces prolonged endoplasmic reticulum stress and autophagic cell death in human umbilical vein endothelial cells. Cardiovasc. Res. 2011, 92, 141–148.
- Zeng, M.; Wei, X.; Wu, Z.; Li, W.; Li, B.; Fei, Y.; He, Y.; Chen, J.; Wang, P.; Liu, X. Reactive oxygen species contribute to simulated ischemia/reperfusion-induced autophagic cell death in human umbilical vein endothelial cells. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2014, 20, 1017–1023.
- Paone, S.; Baxter, A.A.; Hulett, M.D.; Poon, I.K.H. Endothelial cell apoptosis and the role of endothelial cell-derived extracellular vesicles in the progression of atherosclerosis. Cell. Mol. Life Sci. CMLS 2019, 76, 1093–1106.
- Duan, H.; Zhang, Q.; Liu, J.; Li, R.; Wang, D.; Peng, W.; Wu, C. Suppression of apoptosis in vascular endothelial cell, the promising way for natural medicines to treat atherosclerosis. Pharmacol. Res. 2021, 168, 105599.
- Bertheloot, D.; Latz, E.; Franklin, B.S. Necroptosis, pyroptosis and apoptosis: An intricate game of cell death. Cell. Mol. Immunol. 2021, 18, 1106–1121.
- Ketelut-Carneiro, N.; Fitzgerald, K.A. Apoptosis, Pyroptosis, and Necroptosis-Oh My! The Many Ways a Cell Can Die. J. Mol. Biol. 2022, 434, 167378.
- Webster, J.D.; Vucic, D. The Balance of TNF Mediated Pathways Regulates Inflammatory Cell Death Signaling in Healthy and Diseased Tissues. Front. Cell Dev. Biol. 2020, 8, 365.
- Gerrity, R.G.; Richardson, M.; Somer, J.B.; Bell, F.P.; Schwartz, C.J. Endothelial cell morphology in areas of in vivo Evans blue uptake in the aorta of young pigs. II. Ultrastructure of the intima in areas of differing permeability to proteins. Am. J. Pathol. 1977, 89, 313–334.
- Tricot, O.; Mallat, Z.; Heymes, C.; Belmin, J.; Lesèche, G.; Tedgui, A. Relation between endothelial cell apoptosis and blood flow direction in human atherosclerotic plaques. Circulation 2000, 101, 2450–2453.
- Dimmeler, S.; Haendeler, J.; Zeiher, A.M. Regulation of endothelial cell apoptosis in atherothrombosis. Curr. Opin. Lipidol. 2002, 13, 531–536.
- Frey, R.S.; Ushio-Fukai, M.; Malik, A.B. NADPH oxidase-dependent signaling in endothelial cells: Role in physiology and pathophysiology. Antioxid. Redox Signal. 2009, 11, 791–810.
- Qu, K.; Yan, F.; Qin, X.; Zhang, K.; He, W.; Dong, M.; Wu, G. Mitochondrial dysfunction in vascular endothelial cells and its role in atherosclerosis. Front. Physiol. 2022, 13, 1084604.
- Su, Q.; Wang, Y.; Yang, X.; Li, X.D.; Qi, Y.F.; He, X.J.; Wang, Y.J. Inhibition of Endoplasmic Reticulum Stress Apoptosis by Estrogen Protects Human Umbilical Vein Endothelial Cells Through the PI3 Kinase-Akt Signaling Pathway. J. Cell. Biochem. 2017, 118, 4568–4574.
- Gresele, P.; Momi, S.; Guglielmini, G. Nitric oxide-enhancing or -releasing agents as antithrombotic drugs. Biochem. Pharmacol. 2019, 166, 300–312.
- Cai, H.; Harrison, D.G. Endothelial dysfunction in cardiovascular diseases: The role of oxidant stress. Circ. Res. 2000, 87, 840–844.
- Dimmeler, S.; Haendeler, J.; Galle, J.; Zeiher, A.M. Oxidized low-density lipoprotein induces apoptosis of human endothelial cells by activation of CPP32-like proteases. A mechanistic clue to the ‘response to injury’ hypothesis. Circulation 1997, 95, 1760–1763.
- Harada-Shiba, M.; Kinoshita, M.; Kamido, H.; Shimokado, K. Oxidized low density lipoprotein induces apoptosis in cultured human umbilical vein endothelial cells by common and unique mechanisms. J. Biol. Chem. 1998, 273, 9681–9687.
- Sata, M.; Walsh, K. Oxidized LDL activates fas-mediated endothelial cell apoptosis. J. Clin. Investig. 1998, 102, 1682–1689.
- Suc, I.; Escargueil-Blanc, I.; Troly, M.; Salvayre, R.; Nègre-Salvayre, A. HDL and ApoA prevent cell death of endothelial cells induced by oxidized LDL. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 2158–2166.
- Li, D.; Mehta, J.L. Upregulation of endothelial receptor for oxidized LDL (LOX-1) by oxidized LDL and implications in apoptosis of human coronary artery endothelial cells: Evidence from use of antisense LOX-1 mRNA and chemical inhibitors. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1116–1122.
- Dabravolski, S.A.; Sukhorukov, V.N.; Kalmykov, V.A.; Grechko, A.V.; Shakhpazyan, N.K.; Orekhov, A.N. The Role of KLF2 in the Regulation of Atherosclerosis Development and Potential Use of KLF2-Targeted Therapy. Biomedicines 2022, 10, 254.
- Dekker, R.J.; van Soest, S.; Fontijn, R.D.; Salamanca, S.; de Groot, P.G.; VanBavel, E.; Pannekoek, H.; Horrevoets, A.J. Prolonged fluid shear stress induces a distinct set of endothelial cell genes, most specifically lung Krüppel-like factor (KLF2). Blood 2002, 100, 1689–1698.
- Zhang, Y.; Guan, Q.; Wang, Z. PTP1B inhibition ameliorates inflammatory injury and dysfunction in ox-LDL-induced HUVECs by activating the AMPK/SIRT1 signaling pathway via negative regulation of KLF2. Exp. Ther. Med. 2022, 24, 467.
- Li, Q.; Xuan, W.; Jia, Z.; Li, H.; Li, M.; Liang, X.; Su, D. HRD1 prevents atherosclerosis-mediated endothelial cell apoptosis by promoting LOX-1 degradation. Cell Cycle 2020, 19, 1466–1477.
- Lin, K.; Hsu, P.P.; Chen, B.P.; Yuan, S.; Usami, S.; Shyy, J.Y.; Li, Y.S.; Chien, S. Molecular mechanism of endothelial growth arrest by laminar shear stress. Proc. Natl. Acad. Sci. USA 2000, 97, 9385–9389.
- Malek, A.M.; Jiang, L.; Lee, I.; Sessa, W.C.; Izumo, S.; Alper, S.L. Induction of nitric oxide synthase mRNA by shear stress requires intracellular calcium and G-protein signals and is modulated by PI 3 kinase. Biochem. Biophys. Res. Commun. 1999, 254, 231–242.
- Tardy, Y.; Resnick, N.; Nagel, T.; Gimbrone, M.A., Jr.; Dewey, C.F., Jr. Shear stress gradients remodel endothelial monolayers in vitro via a cell proliferation-migration-loss cycle. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 3102–3106.
- Chien, S. Effects of disturbed flow on endothelial cells. Ann. Biomed. Eng. 2008, 36, 554–562.
- Zeng, L.; Zampetaki, A.; Margariti, A.; Pepe, A.E.; Alam, S.; Martin, D.; Xiao, Q.; Wang, W.; Jin, Z.G.; Cockerill, G.; et al. Sustained activation of XBP1 splicing leads to endothelial apoptosis and atherosclerosis development in response to disturbed flow. Proc. Natl. Acad. Sci. USA 2009, 106, 8326–8331.
- Heo, K.S.; Lee, H.; Nigro, P.; Thomas, T.; Le, N.T.; Chang, E.; McClain, C.; Reinhart-King, C.A.; King, M.R.; Berk, B.C.; et al. PKCζ mediates disturbed flow-induced endothelial apoptosis via p53 SUMOylation. J. Cell Biol. 2011, 193, 867–884.
- Birukov, K.G.; Jacobson, J.R.; Flores, A.A.; Ye, S.Q.; Birukova, A.A.; Verin, A.D.; Garcia, J.G. Magnitude-dependent regulation of pulmonary endothelial cell barrier function by cyclic stretch. Am. J. Physiol. Lung Cell. Mol. Physiol. 2003, 285, L785–L797.
- Wang, J.; Fan, B.; Wei, Y.; Suo, X.; Ding, Y. A simple multi-well stretching device to induce inflammatory responses of vascular endothelial cells. Lab A Chip 2016, 16, 360–367.
- Dong, G.; Huang, X.; Jiang, S.; Ni, L.; Chen, S. Simvastatin Mitigates Apoptosis and Transforming Growth Factor-Beta Upregulation in Stretch-Induced Endothelial Cells. Oxidative Med. Cell. Longev. 2019, 2019, 6026051.
- Li, M.; Chiou, K.R.; Bugayenko, A.; Irani, K.; Kass, D.A. Reduced wall compliance suppresses Akt-dependent apoptosis protection stimulated by pulse perfusion. Circ. Res. 2005, 97, 587–595.
- Liu, X.M.; Ensenat, D.; Wang, H.; Schafer, A.I.; Durante, W. Physiologic cyclic stretch inhibits apoptosis in vascular endothelium. FEBS Lett. 2003, 541, 52–56.
- Raaz, U.; Kuhn, H.; Wirtz, H.; Hammerschmidt, S. Rapamycin reduces high-amplitude, mechanical stretch-induced apoptosis in pulmonary microvascular endothelial cells. Microvasc. Res. 2009, 77, 297–303.
- Zhuang, F.; Bao, H.; Shi, Q.; Li, J.; Jiang, Z.L.; Wang, Y.; Qi, Y.X. Endothelial microparticles induced by cyclic stretch activate Src and modulate cell apoptosis. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2020, 34, 13586–13596.
- Ahmadi, Y.; Fard, J.K.; Ghafoor, D.; Eid, A.H.; Sahebkar, A. Paradoxical effects of statins on endothelial and cancer cells: The impact of concentrations. Cancer Cell Int. 2023, 23, 43.
- Sato, K.; Nuki, T.; Gomita, K.; Weyand, C.M.; Hagiwara, N. Statins reduce endothelial cell apoptosis via inhibition of TRAIL expression on activated CD4 T cells in acute coronary syndrome. Atherosclerosis 2010, 213, 33–39.
- Safaeian, L.; Mirian, M.; Bahrizadeh, S. Evolocumab, a PCSK9 inhibitor, protects human endothelial cells against H(2)O(2)-induced oxidative stress. Arch. Physiol. Biochem. 2022, 128, 1681–1686.
- Zeng, J.; Tao, J.; Xi, L.; Wang, Z.; Liu, L. PCSK9 mediates the oxidative low-density lipoprotein-induced pyroptosis of vascular endothelial cells via the UQCRC1/ROS pathway. Int. J. Mol. Med. 2021, 47, 53.
- Hu, Y.; Xu, Q.; Li, H.; Meng, Z.; Hao, M.; Ma, X.; Lin, W.; Kuang, H. Dapagliflozin Reduces Apoptosis of Diabetic Retina and Human Retinal Microvascular Endothelial Cells Through ERK1/2/cPLA2/AA/ROS Pathway Independent of Hypoglycemic. Front. Pharmacol. 2022, 13, 827896.
- Faridvand, Y.; Kazemzadeh, H.; Vahedian, V.; Mirzajanzadeh, P.; Nejabati, H.R.; Safaie, N.; Maroufi, N.F.; Pezeshkian, M.; Nouri, M.; Jodati, A. Dapagliflozin attenuates high glucose-induced endothelial cell apoptosis and inflammation through AMPK/SIRT1 activation. Clin. Exp. Pharmacol. Physiol. 2022, 49, 643–651.
- Yang, J.; Sato, K.; Aprahamian, T.; Brown, N.J.; Hutcheson, J.; Bialik, A.; Perlman, H.; Walsh, K. Endothelial overexpression of Fas ligand decreases atherosclerosis in apolipoprotein E-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1466–1473.
- Singh, R.; Letai, A.; Sarosiek, K. Regulation of apoptosis in health and disease: The balancing act of BCL-2 family proteins. Nat. Rev. Mol. Cell Biol. 2019, 20, 175–193.
- Suresh, K.; Carino, K.; Johnston, L.; Servinsky, L.; Machamer, C.E.; Kolb, T.M.; Lam, H.; Dudek, S.M.; An, S.S.; Rane, M.J.; et al. A nonapoptotic endothelial barrier-protective role for caspase-3. Am. J. Physiol. Lung Cell. Mol. Physiol. 2019, 316, L1118–L1126.
- Avrutsky, M.I.; Ortiz, C.C.; Johnson, K.V.; Potenski, A.M.; Chen, C.W.; Lawson, J.M.; White, A.J.; Yuen, S.K.; Morales, F.N.; Canepa, E.; et al. Endothelial activation of caspase-9 promotes neurovascular injury in retinal vein occlusion. Nat. Commun. 2020, 11, 3173.
- Bader, S.M.; Preston, S.P.; Saliba, K.; Lipszyc, A.; Grant, Z.L.; Mackiewicz, L.; Baldi, A.; Hempel, A.; Clark, M.P.; Peiris, T.; et al. Endothelial Caspase-8 prevents fatal necroptotic hemorrhage caused by commensal bacteria. Cell Death Differ. 2023, 30, 27–36.
- Vandenabeele, P.; Galluzzi, L.; Vanden Berghe, T.; Kroemer, G. Molecular mechanisms of necroptosis: An ordered cellular explosion. Nat. Rev. Mol. Cell Biol. 2010, 11, 700–714.
- Sun, X.; Yin, J.; Starovasnik, M.A.; Fairbrother, W.J.; Dixit, V.M. Identification of a novel homotypic interaction motif required for the phosphorylation of receptor-interacting protein (RIP) by RIP3. J. Biol. Chem. 2002, 277, 9505–9511.
- Petrie, E.J.; Czabotar, P.E.; Murphy, J.M. The Structural Basis of Necroptotic Cell Death Signaling. Trends Biochem. Sci. 2019, 44, 53–63.
- Xia, B.; Fang, S.; Chen, X.; Hu, H.; Chen, P.; Wang, H.; Gao, Z. MLKL forms cation channels. Cell Res. 2016, 26, 517–528.
- Vince, J.E.; Wong, W.W.; Gentle, I.; Lawlor, K.E.; Allam, R.; O’Reilly, L.; Mason, K.; Gross, O.; Ma, S.; Guarda, G.; et al. Inhibitor of apoptosis proteins limit RIP3 kinase-dependent interleukin-1 activation. Immunity 2012, 36, 215–227.
- Karunakaran, D.; Geoffrion, M.; Wei, L.; Gan, W.; Richards, L.; Shangari, P.; DeKemp, E.M.; Beanlands, R.A.; Perisic, L.; Maegdefessel, L.; et al. Targeting macrophage necroptosis for therapeutic and diagnostic interventions in atherosclerosis. Sci. Adv. 2016, 2, e1600224.
- Lin, J.; Li, H.; Yang, M.; Ren, J.; Huang, Z.; Han, F.; Huang, J.; Ma, J.; Zhang, D.; Zhang, Z.; et al. A role of RIP3-mediated macrophage necrosis in atherosclerosis development. Cell Rep. 2013, 3, 200–210.
- Rasheed, A.; Robichaud, S.; Nguyen, M.A.; Geoffrion, M.; Wyatt, H.; Cottee, M.L.; Dennison, T.; Pietrangelo, A.; Lee, R.; Lagace, T.A.; et al. Loss of MLKL (Mixed Lineage Kinase Domain-Like Protein) Decreases Necrotic Core but Increases Macrophage Lipid Accumulation in Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 1155–1167.
- Coornaert, I.; Puylaert, P.; Marcasolli, G.; Grootaert, M.O.J.; Vandenabeele, P.; De Meyer, G.R.Y.; Martinet, W. Impact of myeloid RIPK1 gene deletion on atherogenesis in Apoe-deficient mice. Atherosclerosis 2021, 322, 51–60.
- Newton, K.; Dugger, D.L.; Maltzman, A.; Greve, J.M.; Hedehus, M.; Martin-McNulty, B.; Carano, R.A.; Cao, T.C.; van Bruggen, N.; Bernstein, L.; et al. RIPK3 deficiency or catalytically inactive RIPK1 provides greater benefit than MLKL deficiency in mouse models of inflammation and tissue injury. Cell Death Differ. 2016, 23, 1565–1576.
- Colijn, S.; Muthukumar, V.; Xie, J.; Gao, S.; Griffin, C.T. Cell-specific and athero-protective roles for RIPK3 in a murine model of atherosclerosis. Dis. Models Mech. 2020, 13, dmm041962.
- Karunakaran, D.; Nguyen, M.A.; Geoffrion, M.; Vreeken, D.; Lister, Z.; Cheng, H.S.; Otte, N.; Essebier, P.; Wyatt, H.; Kandiah, J.W.; et al. RIPK1 Expression Associates with Inflammation in Early Atherosclerosis in Humans and Can Be Therapeutically Silenced to Reduce NF-κB Activation and Atherogenesis in Mice. Circulation 2021, 143, 163–177.
- Gan, I.; Jiang, J.; Lian, D.; Huang, X.; Fuhrmann, B.; Liu, W.; Haig, A.; Jevnikar, A.M.; Zhang, Z.X. Mitochondrial permeability regulates cardiac endothelial cell necroptosis and cardiac allograft rejection. Am. J. Transplant. Off. J. Am. Soc. Transplant. Am. Soc. Transpl. Surg. 2019, 19, 686–698.
- Kwok, C.; Pavlosky, A.; Lian, D.; Jiang, J.; Huang, X.; Yin, Z.; Liu, W.; Haig, A.; Jevnikar, A.M.; Zhang, Z.X. Necroptosis Is Involved in CD4+ T Cell-Mediated Microvascular Endothelial Cell Death and Chronic Cardiac Allograft Rejection. Transplantation 2017, 101, 2026–2037.
- Zhu, P.; Wang, J.; Du, W.; Ren, J.; Zhang, Y.; Xie, F.; Xu, G. NR4A1 Promotes LPS-Induced Acute Lung Injury through Inhibition of Opa1-Mediated Mitochondrial Fusion and Activation of PGAM5-Related Necroptosis. Oxidative Med. Cell. Longev. 2022, 2022, 6638244.
- Singla, S.; Sysol, J.R.; Dille, B.; Jones, N.; Chen, J.; Machado, R.F. Hemin Causes Lung Microvascular Endothelial Barrier Dysfunction by Necroptotic Cell Death. Am. J. Respir. Cell Mol. Biol. 2017, 57, 307–314.
- Paku, S.; Laszlo, V.; Dezso, K.; Nagy, P.; Hoda, M.A.; Klepetko, W.; Renyi-Vamos, F.; Timar, J.; Reynolds, A.R.; Dome, B. The evidence for and against different modes of tumour cell extravasation in the lung: Diapedesis, capillary destruction, necroptosis, and endothelialization. J. Pathol. 2017, 241, 441–447.
- Strilic, B.; Yang, L.; Albarrán-Juárez, J.; Wachsmuth, L.; Han, K.; Müller, U.C.; Pasparakis, M.; Offermanns, S. Tumour-cell-induced endothelial cell necroptosis via death receptor 6 promotes metastasis. Nature 2016, 536, 215–218.
- Bolik, J.; Krause, F.; Stevanovic, M.; Gandraß, M.; Thomsen, I.; Schacht, S.S.; Rieser, E.; Müller, M.; Schumacher, N.; Fritsch, J.; et al. Inhibition of ADAM17 impairs endothelial cell necroptosis and blocks metastasis. J. Exp. Med. 2022, 219, e20201039.
- Yu, P.; Zhang, X.; Liu, N.; Tang, L.; Peng, C.; Chen, X. Pyroptosis: Mechanisms and diseases. Signal Transduct. Target. Ther. 2021, 6, 128.
- Coll, R.C.; Schroder, K.; Pelegrín, P. NLRP3 and pyroptosis blockers for treating inflammatory diseases. Trends Pharmacol. Sci. 2022, 43, 653–668.
- Shi, X.; Xie, W.L.; Kong, W.W.; Chen, D.; Qu, P. Expression of the NLRP3 Inflammasome in Carotid Atherosclerosis. J. Stroke Cerebrovasc. Dis. Off. J. Natl. Stroke Assoc. 2015, 24, 2455–2466.
- Paramel Varghese, G.; Folkersen, L.; Strawbridge, R.J.; Halvorsen, B.; Yndestad, A.; Ranheim, T.; Krohg-Sørensen, K.; Skjelland, M.; Espevik, T.; Aukrust, P.; et al. NLRP3 Inflammasome Expression and Activation in Human Atherosclerosis. J. Am. Heart Assoc. 2016, 5, e003031.
- Jin, X.; Fu, W.; Zhou, J.; Shuai, N.; Yang, Y.; Wang, B. Oxymatrine attenuates oxidized low-density lipoprotein-induced HUVEC injury by inhibiting NLRP3 inflammasome-mediated pyroptosis via the activation of the SIRT1/Nrf2 signaling pathway. Int. J. Mol. Med. 2021, 48, 187.
- Qiu, Y.; Li, L.; Guo, X.; Liu, J.; Xu, L.; Li, Y. Exogenous spermine inhibits high glucose/oxidized LDL-induced oxidative stress and macrophage pyroptosis by activating the Nrf2 pathway. Exp. Ther. Med. 2022, 23, 310.
- Duewell, P.; Kono, H.; Rayner, K.J.; Sirois, C.M.; Vladimer, G.; Bauernfeind, F.G.; Abela, G.S.; Franchi, L.; Nuñez, G.; Schnurr, M.; et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 2010, 464, 1357–1361.
- Kayagaki, N.; Stowe, I.B.; Lee, B.L.; O’Rourke, K.; Anderson, K.; Warming, S.; Cuellar, T.; Haley, B.; Roose-Girma, M.; Phung, Q.T.; et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 2015, 526, 666–671.
- Das, B.; Sarkar, C.; Rawat, V.S.; Kalita, D.; Deka, S.; Agnihotri, A. Promise of the NLRP3 Inflammasome Inhibitors in In Vivo Disease Models. Molecules 2021, 26, 4996.
- Zahid, A.; Li, B.; Kombe, A.J.K.; Jin, T.; Tao, J. Pharmacological Inhibitors of the NLRP3 Inflammasome. Front. Immunol. 2019, 10, 2538.
- van der Heijden, T.; Kritikou, E.; Venema, W.; van Duijn, J.; van Santbrink, P.J.; Slütter, B.; Foks, A.C.; Bot, I.; Kuiper, J. NLRP3 Inflammasome Inhibition by MCC950 Reduces Atherosclerotic Lesion Development in Apolipoprotein E-Deficient Mice-Brief Report. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1457–1461.
- Zeng, W.; Wu, D.; Sun, Y.; Suo, Y.; Yu, Q.; Zeng, M.; Gao, Q.; Yu, B.; Jiang, X.; Wang, Y. The selective NLRP3 inhibitor MCC950 hinders atherosclerosis development by attenuating inflammation and pyroptosis in macrophages. Sci. Rep. 2021, 11, 19305.
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131.
- Martinet, W.; Coornaert, I.; Puylaert, P.; De Meyer, G.R.Y. Macrophage Death as a Pharmacological Target in Atherosclerosis. Front. Pharmacol. 2019, 10, 306.
- Qian, Z.; Zhao, Y.; Wan, C.; Deng, Y.; Zhuang, Y.; Xu, Y.; Zhu, Y.; Lu, S.; Bao, Z. Pyroptosis in the Initiation and Progression of Atherosclerosis. Front. Pharmacol. 2021, 12, 652963.
- Lin, X.; Ouyang, S.; Zhi, C.; Li, P.; Tan, X.; Ma, W.; Yu, J.; Peng, T.; Chen, X.; Li, L.; et al. Focus on ferroptosis, pyroptosis, apoptosis and autophagy of vascular endothelial cells to the strategic targets for the treatment of atherosclerosis. Arch. Biochem. Biophys. 2022, 715, 109098.
- Koka, S.; Xia, M.; Chen, Y.; Bhat, O.M.; Yuan, X.; Boini, K.M.; Li, P.L. Endothelial NLRP3 inflammasome activation and arterial neointima formation associated with acid sphingomyelinase during hypercholesterolemia. Redox Biol. 2017, 13, 336–344.
- Wu, X.; Zhang, H.; Qi, W.; Zhang, Y.; Li, J.; Li, Z.; Lin, Y.; Bai, X.; Liu, X.; Chen, X.; et al. Nicotine promotes atherosclerosis via ROS-NLRP3-mediated endothelial cell pyroptosis. Cell Death Dis. 2018, 9, 171.
- Yin, Y.; Li, X.; Sha, X.; Xi, H.; Li, Y.F.; Shao, Y.; Mai, J.; Virtue, A.; Lopez-Pastrana, J.; Meng, S.; et al. Early hyperlipidemia promotes endothelial activation via a caspase-1-sirtuin 1 pathway. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 804–816.
- Zhuang, T.; Liu, J.; Chen, X.; Zhang, L.; Pi, J.; Sun, H.; Li, L.; Bauer, R.; Wang, H.; Yu, Z.; et al. Endothelial Foxp1 Suppresses Atherosclerosis via Modulation of Nlrp3 Inflammasome Activation. Circ. Res. 2019, 125, 590–605.
- Xu, X.; Yang, Y.; Wang, G.; Yin, Y.; Han, S.; Zheng, D.; Zhou, S.; Zhao, Y.; Chen, Y.; Jin, Y. Low shear stress regulates vascular endothelial cell pyroptosis through miR-181b-5p/STAT-3 axis. J. Cell. Physiol. 2021, 236, 318–327.
- Puylaert, P.; Van Praet, M.; Vaes, F.; Neutel, C.H.G.; Roth, L.; Guns, P.J.; De Meyer, G.R.Y.; Martinet, W. Gasdermin D Deficiency Limits the Transition of Atherosclerotic Plaques to an Inflammatory Phenotype in Apoe Knock-Out Mice. Biomedicines 2022, 10, 1171.
- Hu, J.J.; Liu, X.; Xia, S.; Zhang, Z.; Zhang, Y.; Zhao, J.; Ruan, J.; Luo, X.; Lou, X.; Bai, Y.; et al. FDA-approved disulfiram inhibits pyroptosis by blocking gasdermin D pore formation. Nat. Immunol. 2020, 21, 736–745.
- Yang, W.S.; Stockwell, B.R. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem. Biol. 2008, 15, 234–245.
- Liang, D.; Minikes, A.M.; Jiang, X. Ferroptosis at the intersection of lipid metabolism and cellular signaling. Mol. Cell 2022, 82, 2215–2227.
- Stockwell, B.R. Ferroptosis turns 10: Emerging mechanisms, physiological functions, and therapeutic applications. Cell 2022, 185, 2401–2421.
- Seibt, T.M.; Proneth, B.; Conrad, M. Role of GPX4 in ferroptosis and its pharmacological implication. Free Radic. Biol. Med. 2019, 133, 144–152.
- Yang, W.S.; Stockwell, B.R. Ferroptosis: Death by Lipid Peroxidation. Trends Cell Biol. 2016, 26, 165–176.
- Sullivan, J.L. Iron and the sex difference in heart disease risk. Lancet 1981, 1, 1293–1294.
- Kiechl, S.; Willeit, J.; Egger, G.; Poewe, W.; Oberhollenzer, F. Body iron stores and the risk of carotid atherosclerosis: Prospective results from the Bruneck study. Circulation 1997, 96, 3300–3307.
- Meyers, D.G.; Jensen, K.C.; Menitove, J.E. A historical cohort study of the effect of lowering body iron through blood donation on incident cardiac events. Transfusion 2002, 42, 1135–1139.
- Vinchi, F.; Porto, G.; Simmelbauer, A.; Altamura, S.; Passos, S.T.; Garbowski, M.; Silva, A.M.N.; Spaich, S.; Seide, S.E.; Sparla, R.; et al. Atherosclerosis is aggravated by iron overload and ameliorated by dietary and pharmacological iron restriction. Eur. Heart J. 2020, 41, 2681–2695.
- Lee, T.S.; Shiao, M.S.; Pan, C.C.; Chau, L.Y. Iron-deficient diet reduces atherosclerotic lesions in Apoe-deficient mice. Circulation 1999, 99, 1222–1229.
- Minqin, R.; Rajendran, R.; Pan, N.; Tan, B.K.; Ong, W.Y.; Watt, F.; Halliwell, B. The iron chelator desferrioxamine inhibits atherosclerotic lesion development and decreases lesion iron concentrations in the cholesterol-fed rabbit. Free Radic. Biol. Med. 2005, 38, 1206–1211.
- Zhang, W.J.; Wei, H.; Frei, B. The iron chelator, desferrioxamine, reduces inflammation and atherosclerotic lesion development in experimental mice. Exp. Biol. Med. 2010, 235, 633–641.
- Yang, K.; Song, H.; Yin, D. PDSS2 Inhibits the Ferroptosis of Vascular Endothelial Cells in Atherosclerosis by Activating Nrf2. J. Cardiovasc. Pharmacol. 2021, 77, 767–776.
- Bai, T.; Li, M.; Liu, Y.; Qiao, Z.; Wang, Z. Inhibition of ferroptosis alleviates atherosclerosis through attenuating lipid peroxidation and endothelial dysfunction in mouse aortic endothelial cell. Free Radic. Biol. Med. 2020, 160, 92–102.
- Li, L.; Wang, H.; Zhang, J.; Chen, X.; Zhang, Z.; Li, Q. Effect of endothelial progenitor cell-derived extracellular vesicles on endothelial cell ferroptosis and atherosclerotic vascular endothelial injury. Cell Death Discov. 2021, 7, 235.
- Zhao, L.; Yang, N.; Song, Y.; Si, H.; Qin, Q.; Guo, Z. Effect of iron overload on endothelial cell calcification and its mechanism. Ann. Transl. Med. 2021, 9, 1658.
- Döring, Y.; Soehnlein, O.; Weber, C. Neutrophil Extracellular Traps in Atherosclerosis and Atherothrombosis. Circ. Res. 2017, 120, 736–743.
- Mostafa, M.N.; Osama, M. The implications of neutrophil extracellular traps in the pathophysiology of atherosclerosis and atherothrombosis. Exp. Biol. Med. 2020, 245, 1376–1384.
- Döring, Y.; Libby, P.; Soehnlein, O. Neutrophil Extracellular Traps Participate in Cardiovascular Diseases: Recent Experimental and Clinical Insights. Circ. Res. 2020, 126, 1228–1241.
- Nija, R.J.; Sanju, S.; Sidharthan, N.; Mony, U. Extracellular Trap by Blood Cells: Clinical Implications. Tissue Eng. Regen. Med. 2020, 17, 141–153.
- de Boer, O.J.; Li, X.; Teeling, P.; Mackaay, C.; Ploegmakers, H.J.; van der Loos, C.M.; Daemen, M.J.; de Winter, R.J.; van der Wal, A.C. Neutrophils, neutrophil extracellular traps and interleukin-17 associate with the organisation of thrombi in acute myocardial infarction. Thromb. Haemost. 2013, 109, 290–297.
- Döring, Y.; Weber, C.; Soehnlein, O. Footprints of neutrophil extracellular traps as predictors of cardiovascular risk. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1735–1736.
- Gupta, A.K.; Joshi, M.B.; Philippova, M.; Erne, P.; Hasler, P.; Hahn, S.; Resink, T.J. Activated endothelial cells induce neutrophil extracellular traps and are susceptible to NETosis-mediated cell death. FEBS Lett. 2010, 584, 3193–3197.
- Saffarzadeh, M.; Juenemann, C.; Queisser, M.A.; Lochnit, G.; Barreto, G.; Galuska, S.P.; Lohmeyer, J.; Preissner, K.T. Neutrophil extracellular traps directly induce epithelial and endothelial cell death: A predominant role of histones. PLoS ONE 2012, 7, e32366.
- Jiménez-Alcázar, M.; Rangaswamy, C.; Panda, R.; Bitterling, J.; Simsek, Y.J.; Long, A.T.; Bilyy, R.; Krenn, V.; Renné, C.; Renné, T.; et al. Host DNases prevent vascular occlusion by neutrophil extracellular traps. Science 2017, 358, 1202–1206.
- Silvestre-Roig, C.; Braster, Q.; Wichapong, K.; Lee, E.Y.; Teulon, J.M.; Berrebeh, N.; Winter, J.; Adrover, J.M.; Santos, G.S.; Froese, A.; et al. Externalized histone H4 orchestrates chronic inflammation by inducing lytic cell death. Nature 2019, 569, 236–240.
- Franck, G.; Mawson, T.L.; Folco, E.J.; Molinaro, R.; Ruvkun, V.; Engelbertsen, D.; Liu, X.; Tesmenitsky, Y.; Shvartz, E.; Sukhova, G.K.; et al. Roles of PAD4 and NETosis in Experimental Atherosclerosis and Arterial Injury: Implications for Superficial Erosion. Circ. Res. 2018, 123, 33–42.
- Quillard, T.; Araújo, H.A.; Franck, G.; Shvartz, E.; Sukhova, G.; Libby, P. TLR2 and neutrophils potentiate endothelial stress, apoptosis and detachment: Implications for superficial erosion. Eur. Heart J. 2015, 36, 1394–1404.
- Franck, G.; Mawson, T.; Sausen, G.; Salinas, M.; Masson, G.S.; Cole, A.; Beltrami-Moreira, M.; Chatzizisis, Y.; Quillard, T.; Tesmenitsky, Y.; et al. Flow Perturbation Mediates Neutrophil Recruitment and Potentiates Endothelial Injury via TLR2 in Mice: Implications for Superficial Erosion. Circ. Res. 2017, 121, 31–42.
- Molinaro, R.; Yu, M.; Sausen, G.; Bichsel, C.A.; Corbo, C.; Folco, E.J.; Lee, G.Y.; Liu, Y.; Tesmenitsky, Y.; Shvartz, E.; et al. Targeted delivery of protein arginine deiminase-4 inhibitors to limit arterial intimal NETosis and preserve endothelial integrity. Cardiovasc. Res. 2021, 117, 2652–2663.