Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Camila Xu and Version 1 by Abeer Mahmoud.
Anthracycline is identified as one of the cancer treatments most likely to induce a significant decrease in left ventricular (LV) ejection fraction (LVEF), cardiomyopathy, and cardiac ischemia, ultimately leading to heart failure.
- cardiotoxicity
- anthracyclines
- breast cancer
- genetic polymorphism
1. Introduction
Breast cancer is becoming more common and pervasive in the United States, with incidence rates increasing by 0.5% yearly [1]. It ranks second among cancer-related mortality in women, particularly among African American women, who have a higher risk of acquiring breast cancer before age 40 and a higher mortality rate [1]. Although early detection and treatment have reduced mortality, the number of African Americans developing aggressive types of breast cancer continues to rise [2]. The presence or absence of molecular markers for hormone receptors (estrogen and progesterone receptors) and human epidermal growth factor 2 (Her2-neu) differentiates breast cancer subtypes [3]. These molecular subtypes, along with characteristics like the cancer stage, underlying genetic factors, and responsiveness to neoadjuvant therapy before surgery, influence treatment decisions [4].
The most efficient and often used chemotherapy agents for treating breast cancer are those in the anthracycline class [5]. Despite their widespread usage in treating breast cancer, anthracyclines have numerous side effects, on top of which is cardiotoxicity [6]. Anthracycline-induced cardiotoxicity can be either acute or chronic. While acute cardiotoxicity is typically infrequent and dose-independent, chronic anthracycline-induced cardiotoxicity is a more frequent and dose-dependent outcome that eventually promotes the development of heart failure [7]. At the molecular level, anthracyclines promote cardiac muscle damage and endothelial dysfunction by increasing oxidative and nitrosative stress in conjunction with topoisomerase 2-beta inactivation and other molecular alterations discussed in the corresponding section [8].
Despite increased breast cancer survival, cardiovascular disease is becoming more common among survivors [9]. Within the population of breast cancer patients who are 50 and older, cardiovascular disease constitutes 35% of non-cancer-related mortality [9,10][9][10]. The available data derived from the Surveillance, Epidemiology, and End Results (SEER) program, together with other epidemiological studies, reveal that individuals who have survived breast cancer exhibit a higher prevalence of cardiovascular disease compared to women who have not been diagnosed with breast cancer [9,11][9][11]. Additionally, breast cancer survivors had a greater mortality risk from cardiovascular disease than those without cancer [12]. The contribution of anthracyclines to the development and exacerbation of cardiovascular diseases is highlighted in the most recent European Society of Cardiology (ESC) guidelines on cardio-oncology (2022) [13]. Anthracycline is identified as one of the cancer treatments most likely to induce a significant decrease in left ventricular (LV) ejection fraction (LVEF), cardiomyopathy, and cardiac ischemia, ultimately leading to heart failure. Citing over 40 genetic variants associated with anthracycline-induced cardiac toxicity, the ESC guidelines emphasize the significance of taking into consideration numerous variables that can influence the outcome of anthracycline administration. These guidelines also refer to the underrepresentation of minority populations in ongoing clinical trials and emphasize the need to conduct bigger anthracycline-centered trials that include high-risk populations. Moreover, these guidelines are intended to assist oncologists in making informed decisions regarding treatment doses and regimens, preventing the premature cessation of therapy for patients who may benefit from prolonged therapy, and preventing severe complications in patients who are at a greater risk of developing cardiac toxicity. In a nutshell, the ESC guidelines encourage classifying patients based on several factors that determine their risk of anthracycline-induced cardiac toxicity, which is expected to facilitate the early implementation of personalized preventive strategies.
Preexisting cardiac conditions and established cardiovascular risk factors influence the risk of anthracycline-induced cardiotoxicity [14]. African American and Hispanic women are believed to have a higher cardiovascular risk than their White counterparts. A study by Zhang et al. [15] showed a greater incidence of heart failure in breast cancer survivors of African American (12%) and Hispanic heritage (11%) compared to White patients (6%). This situation can be attributed to genetic, environmental, and socioeconomic disparities that exacerbate and contribute to the discrepancies in risk factors [16]. Thus, breast cancer patients from ethnic minorities face an increased risk of anthracycline-induced cardiotoxicity.
Aside from biological risk factors, socioeconomic, environmental, behavioral, and structural disparities all impact overall mortality and morbidity in breast cancer survivors [17]. These factors have been demonstrated to have a more significant impact on racial/ethnic minority groups. Social determinants of health affect not only the incidence of breast cancer but also its progression and survival outcomes, as well as the response to therapy [18]. It is critical to fully comprehend the impact of these factors on the observed discrepancy in cardiotoxicity in response to anthracycline in breast cancer patients.
2. Molecular Mechanisms of Anthracycline-Induced Cardiotoxicity
Anthracyclines have a propensity for mitochondrial accumulation; hence, myocardial tissue is the primary target of anthracyclines due to its high energy demand and, consequently, high mitochondrial density [45][19]. Although cardiomyocytes are the primary target, research has revealed that anthracyclines can also impact other cell types, including fibroblasts, cardiac progenitor cells, and endothelial cells [14,45][14][19]. Anthracyclines produce cardiotoxicity through a wide variety of molecular mechanisms, some of which include the suppression of topoisomerase 2, the production of reactive oxygen species (ROS), alterations in iron metabolism, and changes in Ca2+ signaling [45,46][19][20]. Myofilament degeneration, myocyte loss, mitochondrial enlargement and fragmentation, and vacuolar degeneration of the sarcoplasmic reticulum are some of the typical pathologies that can be detected in the myocardium in response to anthracyclines [47][21]. The myocardium of individuals treated with anthracyclines showed evidence of cell death by apoptosis and necrosis, and consequently, serum troponin levels were raised [48][22]. Here only discuss the most prevalent ones (Figure 1), recognizing that new mechanisms are still being discovered and that the precise mechanism underlying anthracycline-induced cardiotoxicity cannot be determined with certainty.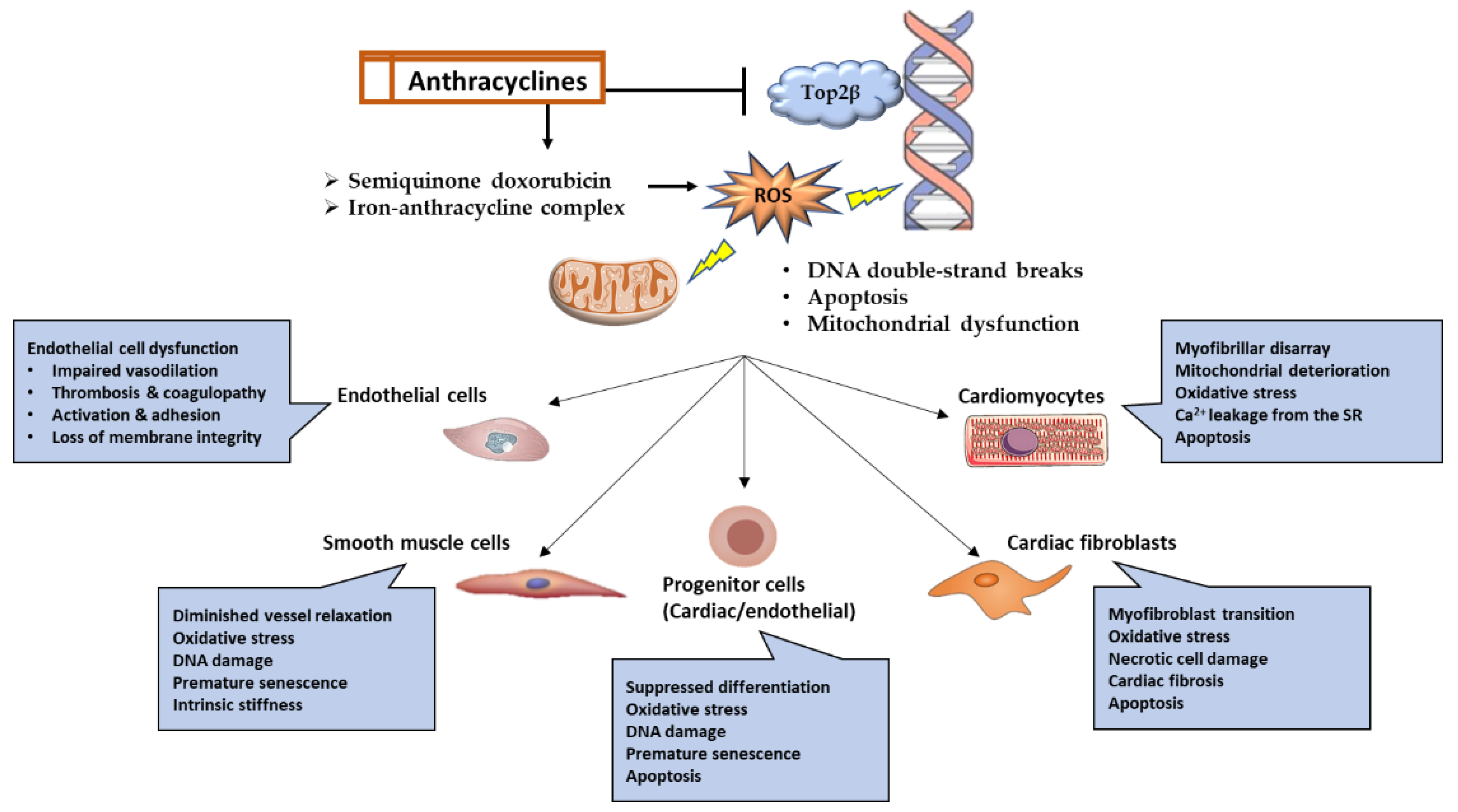
Figure 1. Graphical representation of the molecular pathways and functional outcomes in numerous cell types affected by anthracycline-induced cardiotoxicity.
2.1. Topoisomerase-2 (Top2)
Anthracyclines intercalate into DNA, forming bulky DNA adducts and crosslinks that interfere with the replication and transcription process [7,49][7][23]. Top2 is the main target for doxorubicin, and it has two forms, Top2α, expressed by highly proliferating cells such as cancer cells, and Top2β, expressed by quiescent cells such as cardiomyocytes. Doxorubicin inhibits the catalytic activity of Top2β and stabilizes its intermediary form, causing covalent binding with DNA and persistent DNA double-strand breaks (DSBs). The broken DNA will eventually lead to the recruitment and activation of p53-induced apoptotic cell death. Furthermore, disturbances in the function of Top2β will interfere with the transcription of genes critical for mitochondrial biogenesis, such as peroxisome proliferator-activated receptor gamma receptor co-activators (PPARδ), and mitochondrial function, such as NADH: ubiquinone oxidoreductase subunit A3 (Ndufa3), succinate dehydrogenase complex flavoprotein subunit A (Sdha), and ATP synthase F1 subunit alpha (Atp5a1) [7].2.2. Reactive Oxygen Species
The generation of reactive oxygen species (ROS) is another mechanism through which anthracyclines can damage DNA [7[7][20],46], leading to base mismatch, point mutations, nucleotide oxidation, and DNA single-stranded breaks. Doxorubicin exhibits a selective tendency to accumulate within mitochondria owing to its high affinity for cardiolipin, a phospholipid mostly present in the inner mitochondrial membrane. Doxorubicin can be converted to the semiquinone form via NADPH oxidase and nitric oxide synthase in the cytoplasm and the electron transport chain in the mitochondria. This semiquinone doxorubicin form is unstable and frequently oxidized by oxygen, leading to the generation of significant quantities of ROS and, ultimately, cellular damage and cell death. Mitochondria are abundant in cardiomyocytes, making them a prime target for anthracycline accumulation. Compared to other organs, cardiomyocytes do not exhibit higher levels of antioxidant enzymes. These facts combined demonstrate that cardiomyocytes are highly susceptible to ROS damage caused by anthracyclines [50][24].2.3. Iron Metabolism
Another mechanism by which anthracyclines cause cardiotoxicity is by chelating free iron to form an iron–anthracycline complex. This complex, in turn, reacts with oxygen and promotes ROS production. In addition, anthracyclines interfere with iron metabolism by disrupting iron-binding and iron-transporting proteins. Iron is also sequestered by anthracyclines, interfering with its binding to Iron Regulation Protein-1 (IRP-1). This iron-free portion of IRP-1 promotes transferrin receptor transcription and, as a result, enhances iron uptake by anthracycline-containing cells, particularly the mitochondria-rich cardiomyocytes. In addition to apoptosis and necrosis generated by anthracyclines, the generation of ROS also induces lipid peroxidation and ferroptosis. The latter refers to a recently discovered variant of programmed cell death characterized by the detrimental effects of iron and reactive oxygen species (ROS) on membrane lipids. In support of these findings, recent studies showed that ferroptosis inhibitors could prevent anthracycline-induced cardiotoxicity [50][24].2.4. Guanylate Cyclase Activity
A wide variety of physiological processes important to the function of endothelial cells and vascular smooth muscle cells as well as cardiomyocytes are regulated by the second messenger cyclic guanosine monophosphate (cGMP). Nitric oxide (NO) and carbon monoxide (CO) both activate soluble guanylate cyclase (sGC), which then produces cGMP. Adverse cardiac remodeling and heart failure have been linked to disturbances in the cGMP signaling system. The NO-sGC-cGMP pathway has emerged as a therapeutic target for cardiopulmonary diseases, including heart failure, myocardial ischemia, pulmonary hypertension, and others. These new therapies, which include cinaciguat, riociguat, and vericiguat, have shown promising results in animal models and are currently being evaluated in a number of clinical trials [51][25]. Intriguingly, doxorubicin treatment was found to decrease cardiac cGMP levels, and this has emerged as one of the mechanisms of anthracycline-induced cardiotoxicity [52][26]. Recent studies demonstrated that doxorubicin-induced cardiotoxicity was exacerbated in animal models with reduced cardiac sGC activity [53][27]. In addition, pharmacological administration of sGC improved left ventricular function in animal models receiving doxorubicin compared to the placebo control, indicating the preventive and therapeutic potential of sGC in anthracycline-induced cardiotoxicity [51][25]. Therefore, patients with doxorubicin-induced cardiotoxicity would be ideal candidates for these newly discovered NO-sGC-cGMP-targeting medications.References
- American Cancer Society. Cancer Facts and Figures; American Cancer Society: Atlanta, GA, USA, 2022; Available online: https://www.cancer.org/cancer/breast-cancer/about/how-common-is-breast-cancer.html (accessed on 30 November 2022).
- Yedjou, C.G.; Tchounwou, P.B.; Payton, M.; Miele, L.; Fonseca, D.D.; Lowe, L.; Alo, R.A. Assessing the Racial and Ethnic Disparities in Breast Cancer Mortality in the United States. Int. J. Environ. Res. Public Health 2017, 14, 486.
- Waks, A.G.; Winer, E.P. Breast Cancer Treatment: A Review. JAMA 2019, 321, 288–300.
- Burstein, H.J.; Curigliano, G.; Thurlimann, B.; Weber, W.P.; Poortmans, P.; Regan, M.M.; Senn, H.J.; Winer, E.P.; Gnant, M. Panelists of the St Gallen Consensus Conference. Customizing local and systemic therapies for women with early breast cancer: The St. Gallen International Consensus Guidelines for treatment of early breast cancer 2021. Ann. Oncol. 2021, 32, 1216–1235.
- Venkatesh, P.; Kasi, A. Anthracyclines; StatPearls: Treasure Island, FL, USA, 2022.
- Nicolazzi, M.A.; Carnicelli, A.; Fuorlo, M.; Scaldaferri, A.; Masetti, R.; Landolfi, R.; Favuzzi, A.M.R. Anthracycline and trastuzumab-induced cardiotoxicity in breast cancer. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 2175–2185.
- Sobczuk, P.; Czerwinska, M.; Kleibert, M.; Cudnoch-Jedrzejewska, A. Anthracycline-induced cardiotoxicity and renin-angiotensin-aldosterone system-from molecular mechanisms to therapeutic applications. Heart Fail. Rev. 2022, 27, 295–319.
- Hundley, W.G.; Jordan, J.H. When Left Ventricular Extracellular Volume Fraction Changes After Anthracyclines: Is it Due to a Change in the Numerator, Denominator, or Both? JACC Cardiovasc. Imaging 2018, 11, 1056–1058.
- Gulati, M.; Mulvagh, S.L. The connection between the breast and heart in a woman: Breast cancer and cardiovascular disease. Clin. Cardiol. 2018, 41, 253–257.
- Cousin, L.; Roper, N.; Nolan, T.S. Cardio-Oncology Health Disparities: Social Determinants of Health and Care for Black Breast Cancer Survivors. Clin. J. Oncol. Nurs. 2021, 25, 36–41.
- Bradshaw, P.T.; Stevens, J.; Khankari, N.; Teitelbaum, S.L.; Neugut, A.I.; Gammon, M.D. Cardiovascular Disease Mortality Among Breast Cancer Survivors. Epidemiology 2016, 27, 6–13.
- Dhir, A.A.; Sawant, S.P. Cardiac morbidity & mortality in patients with breast cancer: A review. Indian J. Med. Res. 2021, 154, 199–209.
- Lyon, A.R.; Lopez-Fernandez, T.; Couch, L.S.; Asteggiano, R.; Aznar, M.C.; Bergler-Klein, J.; Boriani, G.; Cardinale, D.; Cordoba, R.; Cosyns, B.; et al. 2022 ESC Guidelines on cardio-oncology developed in collaboration with the European Hematology Association (EHA), the European Society for Therapeutic Radiology and Oncology (ESTRO) and the International Cardio-Oncology Society (IC-OS). Eur. Heart J. 2022, 43, 4229–4361.
- Cardinale, D.; Iacopo, F.; Cipolla, C.M. Cardiotoxicity of Anthracyclines. Front. Cardiovasc. Med. 2020, 7, 26.
- Zhang, L.; Song, J.; Clark, R.; Mangoni, A.; Goldberg, Y.; Slipczuk, L.; Garcia, M.J.; Bansal, N.; Sadler, D.B.; Neilan, T.; et al. Abstract 13090: Racial and Ethnic Differences in Anthracycline Cardiotoxicity. Circulation 2021, 144, A13090.
- Kalinowski, J.; Taylor, J.Y.; Spruill, T.M. Why Are Young Black Women at High Risk for Cardiovascular Disease? Circulation 2019, 139, 1003–1004.
- Zavala, V.A.; Bracci, P.M.; Carethers, J.M.; Carvajal-Carmona, L.; Coggins, N.B.; Cruz-Correa, M.R.; Davis, M.; de Smith, A.J.; Dutil, J.; Figueiredo, J.C.; et al. Cancer health disparities in racial/ethnic minorities in the United States. Br. J. Cancer 2021, 124, 315–332.
- Coughlin, S.S. Social determinants of breast cancer risk, stage, and survival. Breast Cancer Res. Treat. 2019, 177, 537–548.
- Carrasco, R.; Castillo, R.L.; Gormaz, J.G.; Carrillo, M.; Thavendiranathan, P. Role of Oxidative Stress in the Mechanisms of Anthracycline-Induced Cardiotoxicity: Effects of Preventive Strategies. Oxid. Med. Cell Longev. 2021, 2021, 8863789.
- Narezkina, A.; Narayan, H.K.; Zemljic-Harpf, A.E. Molecular mechanisms of anthracycline cardiovascular toxicity. Clin. Sci. 2021, 135, 1311–1332.
- Henriksen, P.A. Anthracycline cardiotoxicity: An update on mechanisms, monitoring and prevention. Heart 2018, 104, 971–977.
- Geisberg, C.A.; Sawyer, D.B. Mechanisms of anthracycline cardiotoxicity and strategies to decrease cardiac damage. Curr. Hypertens. Rep. 2010, 12, 404–410.
- Eom, Y.W.; Kim, M.A.; Park, S.S.; Goo, M.J.; Kwon, H.J.; Sohn, S.; Kim, W.H.; Yoon, G.; Choi, K.S. Two distinct modes of cell death induced by doxorubicin: Apoptosis and cell death through mitotic catastrophe accompanied by senescence-like phenotype. Oncogene 2005, 24, 4765–4777.
- Huang, J.; Wu, R.; Chen, L.; Yang, Z.; Yan, D.; Li, M. Understanding Anthracycline Cardiotoxicity From Mitochondrial Aspect. Front. Pharmacol. 2022, 13, 811406.
- Stasch, J.P.; Pacher, P.; Evgenov, O.V. Soluble guanylate cyclase as an emerging therapeutic target in cardiopulmonary disease. Circulation 2011, 123, 2263–2273.
- Lehotay, D.C.; Levey, B.A.; Rogerson, B.J.; Levey, G.S. Inhibition of cardiac guanylate cyclase by doxorubicin and some of its analogs: Possible relationship to cardiotoxicity. Cancer Treat Rep. 1982, 66, 311–316.
- Vandenwijngaert, S.; Swinnen, M.; Walravens, A.S.; Beerens, M.; Gillijns, H.; Caluwe, E.; Tainsh, R.E.; Nathan, D.I.; Allen, K.; Brouckaert, P.; et al. Decreased Soluble Guanylate Cyclase Contributes to Cardiac Dysfunction Induced by Chronic Doxorubicin Treatment in Mice. Antioxid Redox Signal 2017, 26, 153–164.
More