Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Abeer Mahmoud and Version 2 by Camila Xu.
Anthracycline is identified as one of the cancer treatments most likely to induce a significant decrease in left ventricular (LV) ejection fraction (LVEF), cardiomyopathy, and cardiac ischemia, ultimately leading to heart failure.
- cardiotoxicity
- anthracyclines
- breast cancer
- genetic polymorphism
1. Introduction
Breast cancer is becoming more common and pervasive in the United States, with incidence rates increasing by 0.5% yearly [1]. It ranks second among cancer-related mortality in women, particularly among African American women, who have a higher risk of acquiring breast cancer before age 40 and a higher mortality rate [1]. Although early detection and treatment have reduced mortality, the number of African Americans developing aggressive types of breast cancer continues to rise [2]. The presence or absence of molecular markers for hormone receptors (estrogen and progesterone receptors) and human epidermal growth factor 2 (Her2-neu) differentiates breast cancer subtypes [3]. These molecular subtypes, along with characteristics like the cancer stage, underlying genetic factors, and responsiveness to neoadjuvant therapy before surgery, influence treatment decisions [4].
The most efficient and often used chemotherapy agents for treating breast cancer are those in the anthracycline class [5]. Despite their widespread usage in treating breast cancer, anthracyclines have numerous side effects, on top of which is cardiotoxicity [6]. Anthracycline-induced cardiotoxicity can be either acute or chronic. While acute cardiotoxicity is typically infrequent and dose-independent, chronic anthracycline-induced cardiotoxicity is a more frequent and dose-dependent outcome that eventually promotes the development of heart failure [7]. At the molecular level, anthracyclines promote cardiac muscle damage and endothelial dysfunction by increasing oxidative and nitrosative stress in conjunction with topoisomerase 2-beta inactivation and other molecular alterations discussed in the corresponding section [8].
Despite increased breast cancer survival, cardiovascular disease is becoming more common among survivors [9]. Within the population of breast cancer patients who are 50 and older, cardiovascular disease constitutes 35% of non-cancer-related mortality [9][10][9,10]. The available data derived from the Surveillance, Epidemiology, and End Results (SEER) program, together with other epidemiological studies, reveal that individuals who have survived breast cancer exhibit a higher prevalence of cardiovascular disease compared to women who have not been diagnosed with breast cancer [9][11][9,11]. Additionally, breast cancer survivors had a greater mortality risk from cardiovascular disease than those without cancer [12]. The contribution of anthracyclines to the development and exacerbation of cardiovascular diseases is highlighted in the most recent European Society of Cardiology (ESC) guidelines on cardio-oncology (2022) [13]. Anthracycline is identified as one of the cancer treatments most likely to induce a significant decrease in left ventricular (LV) ejection fraction (LVEF), cardiomyopathy, and cardiac ischemia, ultimately leading to heart failure. Citing over 40 genetic variants associated with anthracycline-induced cardiac toxicity, the ESC guidelines emphasize the significance of taking into consideration numerous variables that can influence the outcome of anthracycline administration. These guidelines also refer to the underrepresentation of minority populations in ongoing clinical trials and emphasize the need to conduct bigger anthracycline-centered trials that include high-risk populations. Moreover, these guidelines are intended to assist oncologists in making informed decisions regarding treatment doses and regimens, preventing the premature cessation of therapy for patients who may benefit from prolonged therapy, and preventing severe complications in patients who are at a greater risk of developing cardiac toxicity. In a nutshell, the ESC guidelines encourage classifying patients based on several factors that determine their risk of anthracycline-induced cardiac toxicity, which is expected to facilitate the early implementation of personalized preventive strategies.
Preexisting cardiac conditions and established cardiovascular risk factors influence the risk of anthracycline-induced cardiotoxicity [14]. African American and Hispanic women are believed to have a higher cardiovascular risk than their White counterparts. A study by Zhang et al. [15] showed a greater incidence of heart failure in breast cancer survivors of African American (12%) and Hispanic heritage (11%) compared to White patients (6%). This situation can be attributed to genetic, environmental, and socioeconomic disparities that exacerbate and contribute to the discrepancies in risk factors [16]. Thus, breast cancer patients from ethnic minorities face an increased risk of anthracycline-induced cardiotoxicity.
Aside from biological risk factors, socioeconomic, environmental, behavioral, and structural disparities all impact overall mortality and morbidity in breast cancer survivors [17]. These factors have been demonstrated to have a more significant impact on racial/ethnic minority groups. Social determinants of health affect not only the incidence of breast cancer but also its progression and survival outcomes, as well as the response to therapy [18]. It is critical to fully comprehend the impact of these factors on the observed discrepancy in cardiotoxicity in response to anthracycline in breast cancer patients.
2. Molecular Mechanisms of Anthracycline-Induced Cardiotoxicity
Anthracyclines have a propensity for mitochondrial accumulation; hence, myocardial tissue is the primary target of anthracyclines due to its high energy demand and, consequently, high mitochondrial density [19][45]. Although cardiomyocytes are the primary target, research has revealed that anthracyclines can also impact other cell types, including fibroblasts, cardiac progenitor cells, and endothelial cells [14][19][14,45]. Anthracyclines produce cardiotoxicity through a wide variety of molecular mechanisms, some of which include the suppression of topoisomerase 2, the production of reactive oxygen species (ROS), alterations in iron metabolism, and changes in Ca2+ signaling [19][20][45,46]. Myofilament degeneration, myocyte loss, mitochondrial enlargement and fragmentation, and vacuolar degeneration of the sarcoplasmic reticulum are some of the typical pathologies that can be detected in the myocardium in response to anthracyclines [21][47]. The myocardium of individuals treated with anthracyclines showed evidence of cell death by apoptosis and necrosis, and consequently, serum troponin levels were raised [22][48]. Here only discuss the most prevalent ones (Figure 1), recognizing that new mechanisms are still being discovered and that the precise mechanism underlying anthracycline-induced cardiotoxicity cannot be determined with certainty.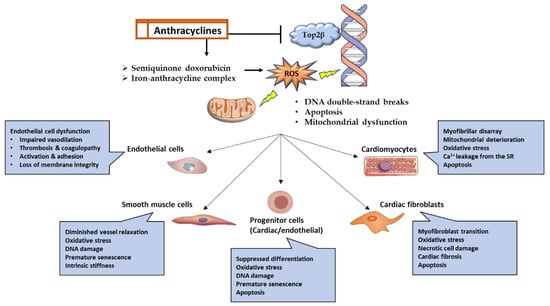
Figure 1. Graphical representation of the molecular pathways and functional outcomes in numerous cell types affected by anthracycline-induced cardiotoxicity.