Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Paulina Miziak and Version 2 by Peter Tang.
Estrogens, belonging to a group of steroid compounds, play an important role in both physiological and disease processes, mainly by interacting with estrogen receptors (ERs). Abnormal ER signaling may result in various cancers, including breast cancer (BC), one of the most often diagnosed cancers in women globally, and a second cause of female cancer-related death.
- estrogen receptor
- ER
- breast cancer
- ER signaling
1. Introduction
One of the most often diagnosed malignancies in women globally is breast cancer (BC), being now the second cause of death because of cancer [1][2][1,2]. The biological activity and treatment response of BC are influenced by a variety of histological and molecular abnormalities [3]. Despite improvements in the development of diagnostic methods and treatments, the incidence and mortality rate of breast cancer-bearing patients are rising internationally [4]. Age, family history, histological differentiation and grading, and the local and systemic advancement of the disease have all been studied to evaluate the patient risk and choose the best course of action [5][6][5,6]. The three main types of breast cancer are classified based on the hormone receptors’ status. The first group consists of tumors that have either tested positive for the estrogen receptor (ER) or the progesterone receptor (PR). The second group consists of tumors that have either tested positive for the human epidermal growth factor receptor 2 (HER2) with or without ER and PR positivity, whereas the third one is called triple-negative breast cancer (TNBC), since these types of tumors lack expression of all three receptors (ER, PR, HER2) [7]. Receptor status, among other variables, has been demonstrated as the one of most important factors in estimating the prognosis and therapeutic response [8]. Furthermore, breast cancer classification based on intrinsic molecular subtypes as a result of the microarray expression profiling has been distinguished [9][10][9,10]. These are termed luminal A (ER+PR+ tumors, expressing luminal genes such as ESR1, GATA3, XBP1, and FOXA1; characterized by the low expression of Ki-67), luminal B (ER+ with lower expression of luminal genes, e.g., PGR and FOX1 and a high expression of Ki-67, >20%), HER2-enriched (characterized by the HER2 positivity; however, not all clinically classified HER+ tumors are of these molecular subtype and intermediate expression of luminal genes), basal-like (increased expression of EGFR and basal cytokeratins with low expression of the luminal A-type genes), and claudin-low (ER-, PR-, and HER- tumors are also negative for claudin 3/4/7 and E-cadherin (reviewed in: [11][12][13][11,12,13]).
ERs are activated by estrogens and play important roles in the development of several cancers; in particular, breast [14], endometrial [15], and ovarian cancers [16]. Estrogens are a group of low molecular weight lipophilic molecules that occur in three forms: estrone (E1), estradiol (E2; the term estrogen is used in relation to E2, due to its predominant role in physiology), and estriol (E3) [17]; the fourth form produced during pregnancy, namely estetrol (E4), is a fetal estrogen with selective tissue actions [18]. These hormones contain in their structure a steroid skeleton made of four aromatic rings. One of them is the phenolic A ring, which is responsible for binding to the ER [19]. Estrogens, like other steroid hormones, are synthesized at the rough endoplasmic reticulum from its precursor—cholesterol, which is described in detail by Fuentes and Silvera (2019) [20]. Briefly, they are synthesized from androstenedione in the presence of oxygen and NADPH. The crucial enzyme involved in this process is aromatase (CYP19A1), an enzyme that participates in the final stage of E1 and E2 synthesis. The synthesis of estrogens takes place in the gonads (predominantly in the ovaries—granulosa cells), adrenal cortex, and adipose tissue, in smaller amounts also in other tissues, including breast and placenta [21], or fetal liver, in the case of E4 [18]. E1 and E2 can arise from testosterone in peripheral tissues (mainly adipose tissue) in the enzymatic reaction catalyzed via aromatase, which has a significant impact on the level of estrogen synthesis in postmenopausal women [22].
Estrogens, including E2 (the predominant circulating estrogen in humans) are transported in the blood along with specific proteins. They sequentially cross biological membranes by diffusing to the target sites, where they primarily act by attaching to specific nuclear ER. Receptor–ligand complexes can directly silence/activate gene expression or act indirectly by interacting with intracellular signaling molecules. The mechanism of action of estrogens is very diverse, and the nature of the response depends on both the genetic and physiological predisposition of the target cells. Estrogens are synthesized in both sexes; however, at different concentrations and with different functions [23]. These hormones play a significant role in the proliferation and growth of cells associated with reproduction and have a myriad of other cellular functions; for instance, carbohydrate and lipid metabolism, and the regulation of energy homeostasis [17][24][17,24]. Importantly, estrogens affect the cardiovascular [25] and central nervous system [26]. The effect of estrogens on the cardiovascular system may be protective, as shown by several studies, including large-scale clinical trials [27][28][29][27,28,29], but have also been associated with the risk of coronary heart disease [30]. Furthermore, estrogen-related malfunctions result in several autoimmune, metabolic, or degenerative pathologies and cancers, including the development of breast cancer [17].
The ER plays a key role in the development, progression, and invasion of ER-expressing BC [31]. ER-positive tumors have a more favorable prognosis compared to other BC types and are usually responsive to hormonal treatment. In the absence of ERα expression, BC exhibits more aggressive phenotypes [32].
2. Estrogen Receptors
The ER family includes the nuclear ER (nER) and G protein-coupled estrogen receptor 1 (GPER1) [33]. nER is characterized by conserved domain structures, such as the DNA-binding domain (DBD) and the ligand-binding domain (LBD) [34]. Two major nER isoforms, ERα and Erβ, are responsible for the regulation of the female reproductive system development, the preservation of bone mass, and the protection of the central nervous system, among other physiologically important processes [35]. The evolutionary origin of the estrogen-signaling system remains unclear; however, the research on invertebrates provided insight into the vertebrate pathway. Interestingly, the ER homologs have been identified in amphioxus [36][37][36,37], mollusks [38][39][38,39], and annelids [40]. Regarding the functional insights, the ERs from amphioxus and mollusks are not activated by estrogens [38][41][42][38,41,42], while in two annelid species, transcription is activated in response to the low doses of estrogens upon ER binding [40]. Based on the phylogenetic context, it was hypothesized the ER possibly originated in the bilateralian lineage [43]. In humans, the nERs are encoded by two different genes (ESR1 for ERα [44] and ESR2 for ERβ [45]) as a result of gene duplication in the early vertebrate lineage [46] that are located on different chromosomes—ESR1 is located on chromosome 6 and ESR2 on chromosome 14. The nER is composed of six homologous A-F domains (Figure 1) representing the receptors’ structural regions and having unique functional characteristics. Domains A and B are located at the amino terminus (N-terminal domain) and contain the so-called activation of function domain 1 (AF-1), whose function is to activate the transcription of target genes [20]. Domain C possesses a zinc-finger motif and corresponds to the DBD domain, namely the DNA-binding domain. This domain is responsible for receptor dimerization and binding to the estrogen-dependent genes promoters’ sequences, called estrogen-response elements (ERE) [47]. The D domain is characterized by the presence of a nuclear localization signal (NLS), which, after the binding of a specific ligand, followed by the conformational change caused by this interaction, is exposed, and it is necessary for translocation to the nucleus. Domain D is the so-called hinge region (H), which is responsible for the functional synergy between fragments AF-1 and the second transcriptional activation domain—the AF-2 fragment located at the carboxyl terminus (C-terminus) [48]. The E domain is the ligand-binding domain (LBD), which contains the ligand-binding site (L). The F domain located at the end of the C-terminus probably acts as a modulator of transcriptional activity and is involved in the interaction with the coactivators [49][50][49,50].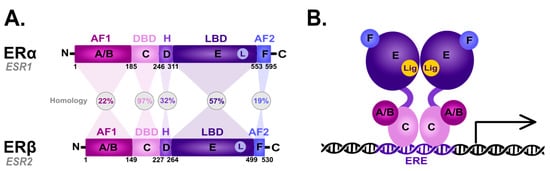
Figure 1. Scheme of the structural and functional regions of the estrogen receptor (ER). (A) Comparison of the domain topology of ERα and ERβ. The homology of the ERα and ERβ receptors was determined based on the amino acid sequence retrieved from the UniProt database (https://www.uniprot.org/; accessed on 12 March 2023; ESR1 ID: P03372, ESR2 ID: Q92731). AF1—the activation of function domain 1; DBD—the DNA-binding domain; H—hinge region; LBD—ligand-binding domain; AF2—the activation of function domain 2; A/B—the domains located at the N-terminus (N); C—the domain containing zinc-finger; D—the domain with nuclear localization signal; E/F—the domains located at the C-terminus (C). (B) Diagram of the estrogen receptor dimer binding to DNA in the estrogen-response elements (ERE). A-F as explained in the description of A; Lig—ligand.
3. Estrogen Signaling
3.1. Genomic Action of ER
The ER-dependent signaling can be classified as genomic and non-genomic with different activities and pathways involved, respectively (Figure 2). Genomic signaling (Figure 2; bottom panel) depends on the transcriptional activities via the gene expression, while non-genomic (Figure 2; top panel) depends on the activation of various signaling cascades, as reviewed in: [20][69][20,69].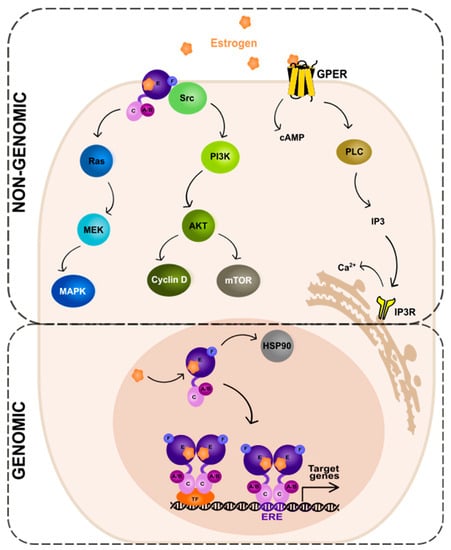
Figure 2. Genomic and non-genomic action of estrogen receptor (ER). Abbreviations: A/B—the domains of ER located at the N-terminus of estrogen receptor (N); C—the domain containing zinc-finger; E/F—the domains located at the C-terminus; GPER—G protein-coupled estrogen receptor 1; PI3K—phosphatidylinositide 3-kinase; AKT—serine/threonine kinase; mTOR—the mammalian target of rapamycin; cAMP—cyclic adenosine monophosphate; PLC—phospholipase C; IP3—inositol trisphosphate; IP3R—inositol trisphosphate receptor; HSP90—heat shock protein 90; ERE—estrogen-response element; TF—transcription factor.