Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Catherine Yang and Version 1 by Diego Franco.
Heart failure constitutes a clinical complex syndrome with different symptomatic characteristics depending on age, sex, race and ethnicity, among others, which has become a major public health issue with an increasing prevalence. One of the most interesting tools seeking to improve prevention, diagnosis, treatment and prognosis of this pathology has focused on finding new molecular biomarkers since heart failure relies on deficient cardiac homeostasis, which is regulated by a strict gene expression. Therefore, currently, analyses of non-coding RNA transcriptomics have been oriented towards human samples.
- heart failure
- transcriptomics
- non-coding RNA
- microRNA
1. Background
Transcriptomic analysis is a comprehensive analysis that provides information about all the RNA transcripts of an organism, including mRNAs and ncRNAs, offering the possibility of measuring gene expression in different developmental stages and physiological/pathological conditions. Transcript studies have been performed for decades. The first approaches used microarrays, in which a large number of genes can be quantitatively detected by using the principle of molecular hybridization [53][1]. Improved techniques were supported by sequence-based approaches [54,55,56][2][3][4] such as Sanger sequencing technique used for random sequences of individual transcripts from cDNA libraries and tag-based methods, i.e., serial analysis of gene expression (SAGE) or cap analysis gene expression (CAGE). However, these technologies are based on the reliance upon existing knowledge about genome sequence. Additional techniques have been introduced to detect unknown genes or alternatively spliced genes. In this sense, RNA-seq technology, which is based on next-generation sequencing (NGS) and whereby shorter reads, allows to sequence thousands or even millions of cDNA molecules at the same time, helping to understand cell function and metabolic mechanisms [57][5]. This methodology has been implemented at the single-cell level (single-cell RNA-seq) identifying the transcriptome and other multi-omic features of different cell types. However, single-cell transcriptomic analyses have some technical limitations such as to recognize lncRNAs only with polyA-tail or low expression levels of this kind of RNA molecules (see Figure 1). Furthermore, most studies using the traditional bulk RNA-seq methods cannot fully characterize the intrinsic heterogeneity between individual cells and the complexity of circRNAs at the single-cell level [58,59][6][7].
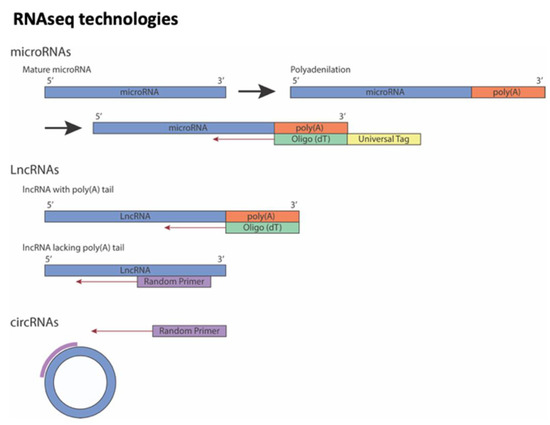
Figure 1. Overview of strategies to sequencing different non-coding RNAs. Note that lncRNAs could be sequencing using Oligo (dT) or Random Primer depending on whether poly(A)-tail on lncRNA is present or not.
2. Transcriptional Analysis of microRNAs in Heart Failure
MicroRNAs are on average 20–22 ribonucleotides in length and display the capacity to bind to the 3’ untranslated region (3´UTR) of coding RNAs by complementary base pairing, promoting their degradation and/or translational blockage. The role of microRNAs as post-transcriptional modulators has been widely described in multiple biological processes, including cell development, differentiation, growth and homeostasis [60,61,62,63][8][9][10][11]. Interestingly, it has been widely described that microRNAs are associated with cardiac physiological and pathological phenotypes, and nowadays circulating microRNAs are being deeply studied as potential HF diagnostic biomarkers.
Particularly, microRNA profiling studies have been carried out in human HF samples, being identified as differentially expressed microRNAs during pathogenesis and progression. In this line, it has been proposed that circulating microRNAs constitute relevant potential biomarkers for the diagnostic of clinical HF [64][12]. By means of microRNA microarray analyses (GSE53437) obtained from HF patients, in plasma and whole blood, 32 microRNAs (validation performed using RT-qPCR) showed expression levels differing from controls. Among these, 12 dysregulated microRNAs were found to be related to specific HF. Even through some of these are specifically related to HFpEF and some others to HPrEF, this is an interesting biomarker tool to distinguish between both types of HF.
In a later study [65][13] concerning microRNA microarray analyses (GSE104150) performed from plasma samples, 94 microRNAs were found to be differentially expressed in patients with clinical HF stage C or D (following the World Health Organization/International Society and the Federation of Cardiology criteria), 7 of them significantly upregulated after RT-qPCR validation.
OurThe comparative analysis (Figure 2) of the above two studies—32 microRNAs (GSE53437) and 94 miRNAs (GSE104150) differentially expressed—presented only two microRNAs in common: miR-320d and miR-671-5p (Figure 2A). When we discarded -5p and -3p ends, four additional microRNAs were observed in common: miR-186, miR-1225, miR-494, and miR-423 (Figure 2B). Finally, when we discarded microRNA isoforms, an extra microRNA appeared in common: miR-92 (Figure 2C). This analysis supports the concept that a limited number of microRNAs are observed in common molecular mechanisms related to HF, while those remaining differentially expressed microRNAs might be attributed to other cofactors such as heterogeneity of the sampling and risk factor index associated with patients.
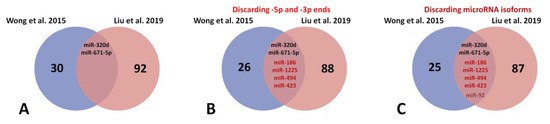
Figure 2. Venn diagram for analysis of commonly deregulated microRNAs related to heart failure in Wong et al., 2015 [64][12] and Liu et al., 2019 [65][13] studies: (A) mir-320d and miR-671 are commonly deregulated microRNAs in HF; (B) if -5p and -3p ends are discarded of the analysis, four new common microRNAs appear to be commonly deregulated; miR-186, miR-1225, miR-494 and miR-423; (C) finally, discarding microRNA isoforms of only one new common microRNA, miR-92, is deregulated in HF.
3. Transcriptomic Analysis of LncRNAs in Heart Failure
LncRNAs [96,97][14][15] are able to function both as transcriptional regulators (modulating nuclear gene expression in different ways, including epigenetic landscape control, transcriptional complex scaffolding and/or decoy molecules) and post-transcriptional regulators (modulating microRNA degradation, mRNA stability and/or protein translation). Notably, several authors have highlighted the pivotal role of lncRNAs in cardiac remodelling and arrhythmogenic pathologies such as dilated cardiomyopathy and atrial fibrillation, respectively, representing HF risk factors [4,19,46,98,99,100][16][17][18][19][20][21]. In this sense, wthe researchers analyzed a number of research studies based on lncRNA transcriptomic approaches performed in samples from both right and left ventricles belonging to HF patients, as described below.
Transcriptomic analyses carried out by monitoring lncRNA alterations of right ventricle (RV) biopsies from HF patients [101][22] reported 78 lncRNAs deregulated in comparison with control samples. Interestingly, 48 of those are represented by classical lncRNAs—35 downregulated and 13 upregulated, while the 30 remaining lncRNAs are catalogued as antisense lncRNAs—18 downregulated and 12 upregulated. Among the first set, lncRNA AP00078783.2 is characterized by displaying the lowest expression levels in HF conditions. Moreover, bioinformatic analysis suggests a potential role of this lncRNA as a microRNA decoy molecule for miR-942, miR-580 and miR-4760-3p [102][23]. In particular, miR-942 has been described as an apoptotic-induced protector in cardiomyocytes. In this sense, by means of in silico analysis, interaction between AP00078783.2 and miR-942 has been observed, suggesting a potential role of this lncRNA in apoptosis regulation. With respect to miR-580 and miR-4760-3p, their possible roles in cardiac pathologies have not been sufficiently explored to date.
The second set includes antisense lncRNAs whose expressions are correlated with the neighboring mRNAs expressions sharing promotor and chromatic features. Within the 30 deregulated antisense lncRNAs [101][22], NPPA-AS1 is characterized by displaying high expression in the HF right ventricle compared to the residual levels found in control samples. Notably, NPPA gene expression is considered as a relevant biomarker in early stages of different cardiac diseases, including HF [103][24]. Similarly to NPPA-AS1, NPPA expression is enhanced in ventricles as a response to cardiac injuries. For this reason, deeper research studies oriented to molecular interaction between both genes are necessary.
Several authors [37][25] have studied left ventricle (LV) appendices to explore possible changes in a transcriptomic environment since this ventricle is the chamber that suffers the most intense remodeling as a consequence of HF. Two transcriptomic analyses of LV samples in HF patients have uncovered multiple lncRNA deregulations as response to this pathological process. Others studies [104][26] have reported differential expression of lncRNAs from LV biopsies of HF induced by ischemic dilated cardiomyopathy. It was observed that 13 lncRNAs were deregulated with respect to control samples—3 upregulated and 10 downregulated. Furthermore, 9 of them—CDKN2B-AS1/ANRIL, EGOT, H19, HOTAIR, LOC285194/TUSC7, RMRP, RNY5, SOX2-OT and SRA1—were detected in blood samples from HF patients, highlighting their potential roles as biomarkers of this disease. By means of Gene Ontology (GO) analysis, these 9 lncRNAs show an association with two biological processes, insulin signaling pathway and cell cycle, which are altered in HF.
Another transcriptomic GO analysis of LV samples from HF patients [105][27] uncovered two additional lncRNAs: AC018647.1 and AC009113.1. Differential expressions of both lncRNAs were detected in three independent transcriptomic datasets. Moreover, competing endogenous RNA (ceRNA) network analysis showed an interrelation between AC018647.1 and AC009113.1, with 170 and 149 associated genes, respectively. ceRNA network analysis suggests that OR51E1 gene is strongly correlated with AC018647.1, whereas RAB9B gene is linked to AC009113.1. Additionally, these two genes have been proposed as modulators of mitochondrial metabolism in HF [106,107][28][29].
Finally, in order to assess possible interactions between lncRNAs and mRNAs in an HF context, several authors [108][30] performed a comprehensive transcriptomic analysis of LV samples from HF. Out of the 993 deregulated lncRNAs found, only 66 of them showed a correlation with a subset of mRNAs. Among those 66 lncRNAs, Neat1 was the most significant, located in the central node of the ceRNA network, displaying the best score. Neat1 expression has also been noticed in cardiac fibrosis and HF, appointed as a relevant biomarker in cardiac disease diagnosis [108,109][30][31].
In tThis review, wee researchers sought to identify if there were any shared lncRNAs as potential biomarkers in HF. The four above studies under analysis did not present any lncRNAs in common (Figure 3). Several reasons could justify these findings: (i) the different regional areas selected by the authors; (ii) the chosen samples obtained from RV vs. LV; (iii) heterogeneous features—age, clinical histories and cardiovascular risk factors—within the cohorts; (iv) variability on the bioinformatic pipelines used to reach stratification and statistical significance of the results, and (v) insufficient sampling data from the transcriptomic analyses available in HF.
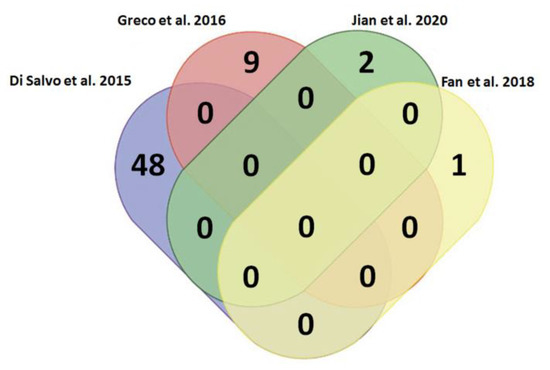
Figure 3. Venn diagram for the analysis of commonly deregulated lncRNAs related to heart failure in Di Salvo et al., 2015 [101][22], Greco et al., 2016 [104][26], Jian et al., 2020 [105][27] and Fan et al., 2018 [108][30]. It has been evidenced that several lncRNAs are deregulated in HF; however, there are not any commonly deregulated among the four studies considered in this review.
4. Transcriptomic Analysis of Circular RNAs in Heart Failure
Circular RNAs (circRNA) display differential expression profiles among species, development stages, and pathologies. Their lack of free ends grants them higher stability in comparison with linear transcripts, becoming attractive candidates as biomarkers and therapeutic tools. Although circRNAs have been described as non-coding RNAs, new discoveries might challenge such consideration, since increasing numbers of studies have found that circRNAs contain open reading frames that can be translated in a cap-independent manner such as internal ribosome entry site (IRES) and N6-methyladenosine (m6A). Furthermore, some peptides generated by circRNAs translation exert physiological function in several tumors such as digestive system neoplasms [28,29,110][32][33][34]. Currently, evidence has been reported on the role of circRNAs in several human diseases, including diabetes mellitus, neurological disorders, chronic inflammatory processes, as well as cardiovascular pathologies [41,111,112,113,114,115,116][35][36][37][38][39][40][41]. The differential expression profiles of circRNAs have been studied in myocardial infarction-induced HF in mice [117,118][42][43]; the main results have been summarized in Figure 4.
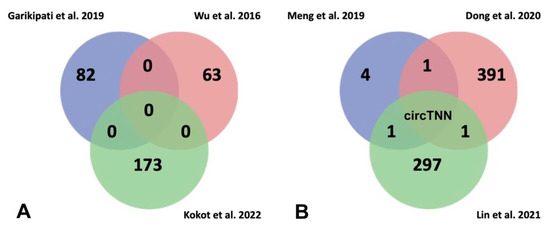
Figure 4. (A) Venn diagram for analysis of commonly deregulated circRNAs related to heart failure in Garikipati et al., 2019 [117][42], Wu et al., 2016 [118][43], and Kokot et al., 2022 [119][44] studies. It has been evidenced that several circRNAs are deregulated in HF; however, there are not any commonly deregulated circRNAs among the three studies considered in this review. (B) Venn diagram for analysis of commonly deregulated circRNAs related to risk factors to heart failure in Meng et al., 2019 [120][45], Dong et al., 2020 [121][46] and Lin et al., 2021 [122][47]. The comparison between them showed that circTTN is deregulated in all studies suggesting a role of possible biomarker of HF.
Several authors have analysed circRNA transcriptomics from myocardial infarction (MI) hearts after 3 days following left anterior descending coronary artery ligation in mice, detecting 82 circRNAs deregulated (41 upregulated and 41 downregulated) compared to sham hearts [117][42]. By means of functional assays, those authors highlighted circFnd3 which attenuates post-MI LV dysfunction modulating positively the endothelial cell function, reducing cardiomyocyte apoptosis and cardiac fibrosis. Furthermore, circFnd3 increased angiogenesis and thus promoted oxygen supply to heart. Although in previous studies [118][43] 63 differentially expressed circRNAs (29 upregulated and 34 downregulated) were identified, no common circRNAs were detected between both studies. Most recently [119][44] it has been demonstrated that adenosine-to-inosine (A-to-I) RNA editing is responsible for 80% of the RNA editing events in human myocardium. Notably, failing hearts display reduced RNA editing, mediated by ADAR2 downregulation which binds to RNA regions and modulates stability of double-stranded RNA and Alu elements. Loss of stability of Alu elements on double-stranded RNA enhance recirculation of pre-mRNAs, resulting in newly formed circRNAs. Deep transcriptomic analysis from failing left ventricle have highlighted misregulation of 173 circRNAs (166 upregulated and 7 downregulated) where the most of upregulated circRNAs are associated with reduced RNA editing in the host gene (Figure 4A). Finally, these authors analyzed the functional role of one of them in hiPSC-CM, circAKAP13, demonstrating that it is essential for sarcomere regularity.
Furthermore, circRNA transcriptomic analyses [120,121,122,123][45][46][47][48] have also been reported in HF risk factor models: (i) in cardiomyocyte hypertrophy, induced by high glucose levels, identifying five differentially expressed circRNAs, and (ii) in dilated cardiomyopathy in two assays performed on human patients, first identifying 392 differentially expressed circRNAs (101 upregulated and 291 downregulated) and second 298 differentially expressed circRNAs (213 upregulated and 85 downregulated). When wthe researhers compared the results of the above works, confluence among them was unappreciated. Only circTTN was identified in two different studies performed in samples from failure human hearts and dilated human hearts, suggesting that this circRNA could be a possible risk biomarker for heart failure (Figure 4B). Therefore, further research should be carried out to consolidate circRNAs as potential HF biomarkers.
References
- Schena, M.; Shalon, D.; Davis, R.W.; Brown, P.O. Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science 1995, 270, 467–470.
- Adams, M.D.; Kelley, J.M.; Gocayne, J.D.; Dubnick, M.A.K.; Polymeropoulos, M.H.; Xiao, H.; Merril, C.R.; Wu, A.; Olde, B.; Moreno, R.F.; et al. Complementary DNA sequencing: Expressed sequence tags and human genome project. Science 1991, 252, 1651–1656.
- Velculescu, V.E.; Zhang, L.; Vogelstein, B.; Kinzler, K.W. Serial analysis of gene expression. Science 1995, 270, 484–487.
- Shiraki, T.; Kondo, S.; Katayama, S.; Waki, K.; Kasukawa, T.; Kawaji, H.; Kodzius, R.; Watahiki, A.; Nakamura, M.; Arakawa, T.; et al. Cap analysis gene expression for high-throughput analysis of transcriptional starting point and identification of promoter usage. Proc. Natl. Acad. Sci. USA 2003, 100, 15776–15781.
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63.
- Li, Y.; Xu, Q.; Wu, D.; Chen, G. Exploring additional valuable information from single-cell RNA-Seq data. Front. Cell Dev. Biol. 2020, 8, 593007.
- Wu, W.; Zhang, J.; Cao, X.; Cai, Z.; Zhao, F. Exploring the cellular landscape of circular RNAs using full-length single-cell RNA sequencing. Nat. Commun. 2022, 13, 3242.
- Lu, M.; Zhang, Q.; Deng, M.; Miao, J.; Guo, Y.; Gao, W.; Cui, Q. An analysis of human microRNA and disease associations. PLoS ONE 2008, 3, e3420.
- Singh, S.R.; Rameshwar, P. MicroRNA in Development and in the Progression of Cancer; Springer: New York, NY, USA, 2014.
- Garcia-Padilla, C.; Garcia-Lopez, V.; Aranega, A.; Franco, D.; Garcia-Martinez, V.; Lopez-Sanchez, C. Inhibition of RhoA and Cdc42 by miR-133a modulates retinoic acid signalling during early development of posterior cardiac tube segment. Int. J. Mol. Sci. 2022, 23, 4179.
- Garcia-Padilla, C.; Dueñas, A.; Franco, D.; Garcia-Lopez, V.; Aranega, A.; Garcia-Martinez, V.; Lopez-Sanchez, C. Dynamic microRNA expression profiles during embryonic development provide novel insights into cardiac sinus venosus/inflow tract differentiation. Front. Cell Dev. Biol. 2022, 9, 767954.
- Wong, L.L.; Armugam, A.; Sepramaniam, S.; Karolina, D.S.; Lim, K.Y.; Lim, J.Y.; Chong, J.P.; Ng, J.Y.; Chen, Y.T.; Chan, M.M.; et al. Circulating microRNAs in heart failure with reduced and preserved left ventricular ejection fraction. Eur. J. Heart Fail. 2015, 17, 393–404.
- Liu, W.; Zheng, J.; Dong, J.; Bai, R.; Song, D.; Ma, X.; Zhao, L.; Yao, Y.; Zhang, H.; Liu, T. Association of miR-197-5p, a circulating biomarker for heart failure, with myocardial fibrosis and adverse cardiovascular events among patients with stage C or D heart failure. Cardiology 2019, 141, 212–225.
- Mathieu, E.L.; Belhocine, M.; Dao, L.T.; Puthier, D.; Spicuglia, S. Rôle des longs ARN non codants dans le développement normal et pathologique . Med. Sci. 2014, 30, 790–796. (In French)
- Engreitz, J.M.; Haines, J.E.; Perez, E.M.; Munson, G.; Chen, J.; Kane, M.; McDonel, P.E.; Guttman, M.; Lander, E.S. Local regulation of gene expression by lncRNA promoters, transcription and splicing. Nature 2016, 539, 452–455.
- Ziaeian, B.; Fonarow, G.C. Epidemiology and aetiology of heart failure. Nat. Rev. Cardiol. 2016, 13, 368–378.
- Gomes, C.P.C.; Schroen, B.; Kuster, G.M.; Robinson, E.L.; Ford, K.; Squire, I.B.; Heymans, S.; Martelli, F.; Emanueli, C.; Devaux, Y. EU-CardioRNA COST Action (CA17129). Regulatory RNAs in Heart Failure. Circulation 2020, 141, 313–328.
- Han, P.; Li, W.; Lin, C.-H.; Yang, J.; Shang, C.; Nurnberg, S.T. A long Noncoding RNA protects the heart from pathological hypertrophy. Nature 2014, 514, 102–106.
- Lozano-Velasco, E.; Garcia-Padilla, C.; Aránega, A.E.; Franco, D. Genetics of atrial fibrilation: In search of novel therapeutic targets. Cardiovasc. Hematol. Disord. Drug Targets 2019, 19, 183–194.
- Rosenbaum, A.N.; Agre, K.E.; Pereira, N.L. Genetics of dilated cardiomyopathy: Practical implications for heart failure management. Nat. Rev. Cardiol. 2020, 17, 286–297.
- Yuan, Z.; Huang, W. New developments in exosomal lncRNAs in cardiovascular diseases. Front. Cardiovasc. Med. 2021, 8, 709169.
- Di Salvo, T.G.; Guo, Y.; Su, Y.R.; Clark, T.; Brittain, E.; Absi, T.; Maltais, S.; Hemnes, A. Right ventricular long noncoding RNA expression in human heart failure. Pulm. Circ. 2015, 5, 135–161.
- Wang, H.; Lin, X.; Li, J.; Zeng, G.; Xu, T. Long Noncoding RNA SOX2-OT aggravates doxorubicin-induced apoptosis of cardiomyocyte by targeting miR-942-5p/DP5. Drug Des. Devel. Ther. 2021, 15, 481–492.
- Man, J.; Barnett, P.; Christoffels, V.M. Structure and function of the Nppa-Nppb cluster locus during heart development and disease. Cell Mol. Life Sci. 2018, 75, 1435–1444.
- Tham, Y.K.; Bernardo, B.C.; Ooi, J.Y.; Weeks, K.L.; McMullen, J.R. Pathophysiology of cardiac hypertrophy and heart failure: Signaling pathways and novel therapeutic targets. Arch. Toxicol. 2015, 89, 1401–1438.
- Greco, S.; Zaccagnini, G.; Perfetti, A.; Fuschi, P.; Valaperta, R.; Voellenkle, C.; Castelvecchio, S.; Gaetano, C.; Finato, N.; Beltrami, A.P.; et al. Long noncoding RNA dysregulation in ischemic heart failure. J. Transl. Med. 2016, 14, 183.
- Jiang, F.; Fan, H.; Luo, L.; Li, Y. An integrative transcriptome analysis reveals consistently dysregulated Long Noncoding RNAs and their transcriptional regulation relationships in heart failure. J. Comput. Biol. 2020, 27, 958–964.
- Liang, Q.; Kobayashi, S. Mitochondrial quality control in the diabetic heart. J. Mol. Cell Cardiol. 2016, 95, 57–69.
- Jovancevic, N.; Dendorfer, A.; Matzkies, M.; Kovarova, M.; Heckmann, J.C.; Osterloh, M.; Boehm, M.; Weber, L.; Nguemo, F.; Semmler, J.; et al. Medium-chain fatty acids modulate myocardial function via a cardiac odorant receptor. Basic Res. Cardiol. 2017, 112, 13.
- Fan, Z.; Gao, S.; Chen, Y.; Xu, B.; Yu, C.; Yue, M.; Tan, X. Integrative analysis of competing endogenous RNA networks reveals the functional lncRNAs in heart failure. J. Cell Mol. Med. 2018, 22, 4818–4829.
- Ge, Z.; Yin, C.; Li, Y.; Tian, D.; Xiang, Y.; Li, Q.; Tang, Y.; Zhang, Y. Long noncoding RNA NEAT1 promotes cardiac fibrosis in heart failure through increased recruitment of EZH2 to the Smad7 promoter region. J. Transl. Med. 2022, 20, 7.
- Li, X.; Yang, L.; Chen, L.L. The biogenesis, functions, and challenges of circular RNAs. Mol. Cell 2018, 71, 428–442.
- Kristensen, L.S.; Andersen, M.S.; Stagsted, L.V.W.; Ebbesen, K.K.; Hansen, T.B.; Kjems, J. The biogenesis, biology and characterization of circular RNAs. Nat. Rev. Genet. 2019, 20, 675–691.
- Meng, E.; Deng, J.; Jiang, R.; Wu, H. CircRNA-encoded peptides or proteins as new players in digestive system neoplasms. Front. Oncol. 2022, 12, 944159.
- Altesha, M.A.; Ni, T.; Khan, A.; Liu, K.; Zheng, X. Circular RNA in cardiovascular disease. J. Cell Physiol. 2019, 234, 5588–5600.
- Holdt, L.M.; Stahringer, A.; Sass, K.; Pichler, G.; Kulak, N.A.; Wilfert, W.; Kohlmaier, A.; Herbst, A.; Northoff, B.H.; Nicolaou, A.; et al. Circular non-coding RNA ANRIL modulates ribosomal RNA maturation and atherosclerosis in humans. Nat. Commun 2016, 7, 12429.
- Devaux, Y.; Creemers, E.E.; Boon, R.A.; Werfel, S.; Thum, T.; Engelhardt, S.; Dimmeler, S.; Squire, I. Cardiolinc network. Circular RNAs in heart failure. Eur. J. Heart Fail. 2017, 9, 701–709.
- Hanan, M.; Soreq, H.; Kadener, S. CircRNAs in the brain. RNA Biol. 2017, 14, 1028–1034.
- Kristensen, L.S.; Hansen, T.B.; Venø, M.T.; Kjems, J. Circular RNAs in cancer: Opportunities and challenges in the field. Oncogene 2018, 37, 555–565.
- Fang, Y.; Wang, X.; Li, W.; Han, J.; Jin, J.; Su, F.; Zhang, J.; Huang, W.; Xiao, F.; Pan, Q.; et al. Screening of circular RNAs and validation of circANKRD36 associated with inflammation in patients with type 2 diabetes mellitus. Int. J. Mol. Med. 2018, 42, 1865–1874.
- Wang, Y.; Liu, B. Circular RNA in Diseased Heart. Cells 2020, 9, 1240.
- Garikipati, V.N.S.; Verma, S.K.; Cheng, Z.; Liang, D.; Truongcao, M.M.; Cimini, M.; Yue, Y.; Huang, G.; Wang, C.; Benedict, C.; et al. Circular RNA CircFndc3b modulates cardiac repair after myocardial infarction via FUS/VEGF-A axis. Nat. Commun. 2020, 10, 4317, Erratum in Nat. Commun. 2020, 11, 2234.
- Wu, H.J.; Zhang, C.Y.; Zhang, S.; Chang, M.; Wang, H.Y. Microarray expression profile of circular RNAs in heart tissue of mice with myocardial infarction-induced heart failure. Cell Physio. Biochem. 2016, 39, 205–216.
- Kokot, K.E.; Kneuer, J.M.; John, D.; Rebs, S.; Möbius-Winkler, M.N.; Erbe, S.; Müller, M.; Andritschke, M.; Gaul, S.; Sheikh, B.N.; et al. Reduction of A-to-I RNA editing in the failing human heart regulates formation of circular RNAs. Basic Res. Cardiol. 2022, 117, 32.
- Meng, Z.; Chen, C.; Cao, H.; Wang, J.; Shen, E. Whole transcriptome sequencing reveals biologically significant RNA markers and related regulating biological pathways in cardiomyocyte hypertrophy induced by high glucose. J. Cell Biochem. 2019, 120, 1018–1027.
- Dong, K.; He, X.; Su, H.; Fulton, D.J.R.; Zhou, J. Genomic analysis of circular RNAs in heart. BMC Med. Genom. 2020, 13, 167.
- Lin, Z.; Zhao, Y.; Dai, F.; Su, E.; Li, F.; Yan, Y. Analysis of changes in circular RNA expression and construction of ceRNA networks in human dilated cardiomyopathy. J. Cell. Mol. Med. 2021, 25, 2572–2583.
- Khan, M.A.; Reckman, Y.J.; Aufiero, S.; van den Hoogenhof, M.M.; van der Made, I.; Beqqali, A.; Koolbergen, D.R.; Rasmussen, T.B.; van der Velden, J.; Creemers, E.E.; et al. RBM20 regulates circular RNA production from the Titin gene. Circ. Res. 2016, 119, 996–1003.
More