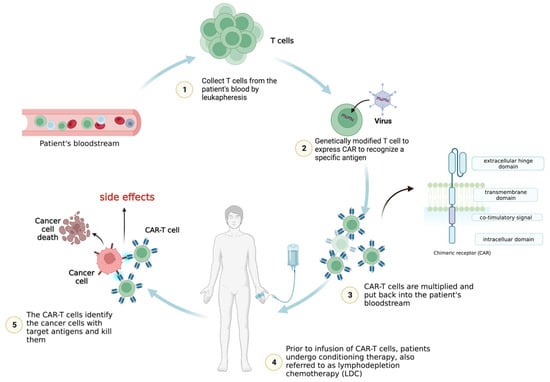
T cells can be genetically engineered to host chimeric antigen receptors (CARs), which can enable the identification and elimination of cancer cells. CARs usually consist of a single-chain variable fragment (scFv, extracellular ligand-binding domain), a spacer domain, a transmembrane region, and intracellular domains. Numerous CAR-T cell treatments have demonstrated exceptional clinical success in treating hematologic malignancies and displayed the immense promise of this ground-breaking technique for cancer immunotherapy. The main impediments to the development of CAR-T cell therapies are the following: cytokine release syndrome (CRS), immune-effector-cell-associated neurotoxicity syndrome (ICANS), tumor lysis syndrome (TLS), and on-target/off-tumor toxicity (OTOT).
- CAR-T cell therapy
- cytokine release syndrome
- immune-effector-cell-associated neurotoxicity syndrome
- tumor lysis syndrome
- OTOT
- toxicity
1. Introduction
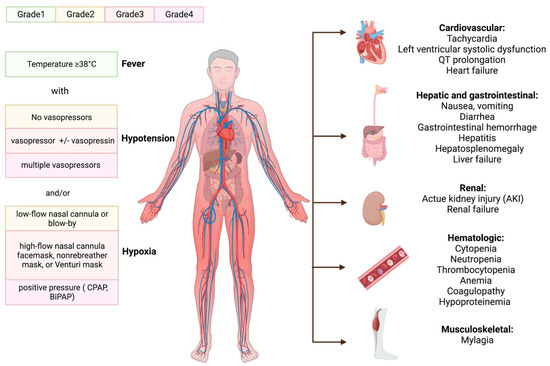
2. Cytokine Release Syndrome (CRS)

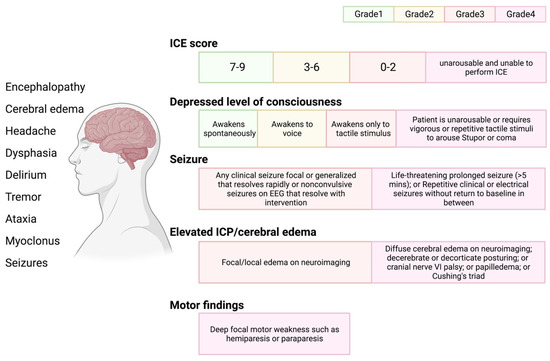
3. Immune-Effector-Cell-Associated Neurotoxicity Syndrome (ICANS)
4. Tumor Lysis Syndrome (TLS)
5. On-Target, Off-Tumor Toxicity (OTOT)
6. Additional Factors Associated with Toxicity
References
- Gill, S.; Maus, M.V.; Porter, D.L. Chimeric Antigen Receptor T Cell Therapy: 25years in the Making. Blood Rev. 2016, 30, 157–167.
- Fesnak, A.D.; June, C.H.; Levine, B.L. Engineered T Cells: The Promise and Challenges of Cancer Immunotherapy. Nat. Rev. Cancer 2016, 16, 566–581.
- Raje, N.; Berdeja, J.; Lin, Y.; Siegel, D.; Jagannath, S.; Madduri, D.; Liedtke, M.; Rosenblatt, J.; Maus, M.V.; Turka, A.; et al. Anti-BCMA CAR T-Cell Therapy Bb2121 in Relapsed or Refractory Multiple Myeloma. N. Engl. J. Med. 2021, 380, 1726–1737.
- Wei, J.; Guo, Y.; Wang, Y.; Wu, Z.; Bo, J.; Zhang, B.; Zhu, J.; Han, W. Clinical Development of CAR T Cell Therapy in China: 2020 Update. Cell Mol. Immunol. 2021, 18, 792–804.
- El-Khazragy, N.; Ghozy, S.; Emad, P.; Mourad, M.; Razza, D.; Farouk, Y.K.; Mohamed, N.A.; Ahmed, M.K.; Youssef, T.; Bahnasawy, Y.M.; et al. Chimeric Antigen Receptor T Cells Immunotherapy: Challenges and Opportunities in Hematological Malignancies. Immunotherapy 2020, 12, 1341–1357.
- Xiao, X.; Huang, S.; Chen, S.; Wang, Y.; Sun, Q.; Xu, X.; Li, Y. Mechanisms of Cytokine Release Syndrome and Neurotoxicity of CAR T-Cell Therapy and Associated Prevention and Management Strategies. J. Exp. Clin. Cancer Res. 2021, 40, 367.
- Brentjens, R.; Yeh, R.; Bernal, Y.; Riviere, I.; Sadelain, M. Treatment of Chronic Lymphocytic Leukemia with Genetically Targeted Autologous T Cells: Case Report of an Unforeseen Adverse Event in a Phase I Clinical Trial. Mol. Ther. 2010, 18, 666–668.
- Grupp, S.A.; Kalos, M.; Barrett, D.; Aplenc, R.; Porter, D.L.; Rheingold, S.R.; Teachey, D.T.; Chew, A.; Hauck, B.; Wright, J.F.; et al. Chimeric Antigen Receptor–Modified T Cells for Acute Lymphoid Leukemia. N. Engl. J. Med. 2013, 368, 1509–1518.
- Morgan, R.A.; Yang, J.C.; Kitano, M.; Dudley, M.E.; Laurencot, C.M.; Rosenberg, S.A. Case Report of a Serious Adverse Event Following the Administration of T Cells Transduced with a Chimeric Antigen Receptor Recognizing ERBB2. Mol. Ther. 2010, 18, 843–851.
- Bullock, T.N.J. CD40 Stimulation as a Molecular Adjuvant for Cancer Vaccines and Other Immunotherapies. Cell Mol. Immunol. 2022, 19, 14–22.
- Tang, T.; Cheng, X.; Truong, B.; Sun, L.; Yang, X.; Wang, H. Molecular Basis and Therapeutic Implications of CD40/CD40L Immune Checkpoint. Pharmacol. Ther. 2021, 219, 107709.
- Singh, N.; Hofmann, T.J.; Gershenson, Z.; Levine, B.L.; Grupp, S.A.; Teachey, D.T.; Barrett, D.M. Monocyte Lineage-Derived IL-6 Does Not Affect Chimeric Antigen Receptor T-Cell Function. Cytotherapy 2017, 19, 867–880.
- Interleukin-6: Designing Specific Therapeutics for a Complex Cytokine. Available online: https://pubmed.ncbi.nlm.nih.gov/29725131/ (accessed on 6 April 2023).
- Teachey, D.T.; Lacey, S.F.; Shaw, P.A.; Melenhorst, J.J.; Maude, S.L.; Frey, N.; Pequignot, E.; Gonzalez, V.E.; Chen, F.; Finklestein, J.; et al. Identification of Predictive Biomarkers for Cytokine Release Syndrome after Chimeric Antigen Receptor T-Cell Therapy for Acute Lymphoblastic Leukemia. Cancer Discov. 2016, 6, 664–679.
- Zhang, X.; Zhu, L.; Zhang, H.; Chen, S.; Xiao, Y. CAR-T Cell Therapy in Hematological Malignancies: Current Opportunities and Challenges. Front. Immunol. 2022, 13, 927153.
- Granzyme a from Cytotoxic Lymphocytes Cleaves GSDMB to Trigger Pyroptosis in Target Cells. Available online: https://pubmed.ncbi.nlm.nih.gov/32299851/ (accessed on 6 April 2023).
- Gasdermin E-Mediated Target Cell Pyroptosis by CAR T Cells Triggers Cytokine Release Syndrome. Available online: https://pubmed.ncbi.nlm.nih.gov/31953257/ (accessed on 6 April 2023).
- Gong, T.; Liu, L.; Jiang, W.; Zhou, R. DAMP-Sensing Receptors in Sterile Inflammation and Inflammatory Diseases. Nat. Rev. Immunol. 2020, 20, 95–112.
- Lee, D.W.; Gardner, R.; Porter, D.L.; Louis, C.U.; Ahmed, N.; Jensen, M.; Grupp, S.A.; Mackall, C.L. Current Concepts in the Diagnosis and Management of Cytokine Release Syndrome. Blood 2014, 124, 188–195.
- Brudno, J.N.; Kochenderfer, J.N. Toxicities of Chimeric Antigen Receptor T Cells: Recognition and Management. Blood 2016, 127, 3321–3330.
- Brudno, J.N.; Kochenderfer, J.N. Recent Advances in CAR T-Cell Toxicity: Mechanisms, Manifestations and Management. Blood Rev. 2019, 34, 45–55.
- Freyer, C.W.; Porter, D.L. Cytokine Release Syndrome and Neurotoxicity Following CAR T-Cell Therapy for Hematologic Malignancies. J. Allergy Clin. Immunol. 2020, 146, 940–948.
- Brentjens, R.J.; Rivière, I.; Park, J.H.; Davila, M.L.; Wang, X.; Stefanski, J.; Taylor, C.; Yeh, R.; Bartido, S.; Borquez-Ojeda, O.; et al. Safety and Persistence of Adoptively Transferred Autologous CD19-Targeted T Cells in Patients with Relapsed or Chemotherapy Refractory B-Cell Leukemias. Blood 2011, 118, 4817–4828.
- Lee, D.W.; Kochenderfer, J.N.; Stetler-Stevenson, M.; Cui, Y.K.; Delbrook, C.; Feldman, S.A.; Fry, T.J.; Orentas, R.; Sabatino, M.; Shah, N.N.; et al. T Cells Expressing CD19 Chimeric Antigen Receptors for Acute Lymphoblastic Leukaemia in Children and Young Adults: A Phase 1 Dose-Escalation Trial. Lancet 2015, 385, 517–528.
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric Antigen Receptor T Cells for Sustained Remissions in Leukemia. N. Engl. J. Med. 2014, 371, 1507–1517.
- Porter, D.L.; Hwang, W.-T.; Frey, N.V.; Lacey, S.F.; Shaw, P.A.; Loren, A.W.; Bagg, A.; Marcucci, K.T.; Shen, A.; Gonzalez, V.; et al. Chimeric Antigen Receptor T Cells Persist and Induce Sustained Remissions in Relapsed Refractory Chronic Lymphocytic Leukemia. Sci. Transl. Med. 2015, 7, 303ra139.
- Wang, Y.; Zhang, W.; Han, Q.; Liu, Y.; Dai, H.; Guo, Y.; Bo, J.; Fan, H.; Zhang, Y.; Zhang, Y.; et al. Effective Response and Delayed Toxicities of Refractory Advanced Diffuse Large B-Cell Lymphoma Treated by CD20-Directed Chimeric Antigen Receptor-Modified T Cells. Clin. Immunol. 2014, 155, 160–175.
- Turtle, C.J.; Hanafi, L.-A.; Berger, C.; Gooley, T.A.; Cherian, S.; Hudecek, M.; Sommermeyer, D.; Melville, K.; Pender, B.; Budiarto, T.M.; et al. CD19 CAR–T Cells of Defined CD4+:CD8+ Composition in Adult B Cell ALL Patients. J. Clin. Investig. 2016, 126, 2123–2138.
- Fried, S.; Avigdor, A.; Bielorai, B.; Meir, A.; Besser, M.J.; Schachter, J.; Shimoni, A.; Nagler, A.; Toren, A.; Jacoby, E. Early and Late Hematologic Toxicity Following CD19 CAR-T Cells. Bone Marrow Transplant. 2019, 54, 1643–1650.
- Brudno, J.N.; Somerville, R.P.T.; Shi, V.; Rose, J.J.; Halverson, D.C.; Fowler, D.H.; Gea-Banacloche, J.C.; Pavletic, S.Z.; Hickstein, D.D.; Lu, T.L.; et al. Allogeneic T Cells That Express an Anti-CD19 Chimeric Antigen Receptor Induce Remissions of B-Cell Malignancies That Progress After Allogeneic Hematopoietic Stem-Cell Transplantation Without Causing Graft-Versus-Host Disease. J. Clin. Oncol. 2016, 34, 1112–1121.
- Ali, S.A.; Shi, V.; Maric, I.; Wang, M.; Stroncek, D.F.; Rose, J.J.; Brudno, J.N.; Stetler-Stevenson, M.; Feldman, S.A.; Hansen, B.G.; et al. T Cells Expressing an Anti-B-Cell Maturation Antigen Chimeric Antigen Receptor Cause Remissions of Multiple Myeloma. Blood 2016, 128, 1688–1700.
- Jensen, M.C.; Popplewell, L.; Cooper, L.J.; DiGiusto, D.; Kalos, M.; Ostberg, J.R.; Forman, S.J. Antitransgene Rejection Responses Contribute to Attenuated Persistence of Adoptively Transferred CD20/CD19-Specific Chimeric Antigen Receptor Redirected T Cells in Humans. Biol. Blood Marrow Transplant. 2010, 16, 1245–1256.
- Kochenderfer, J.N.; Dudley, M.E.; Feldman, S.A.; Wilson, W.H.; Spaner, D.E.; Maric, I.; Stetler-Stevenson, M.; Phan, G.Q.; Hughes, M.S.; Sherry, R.M.; et al. B-Cell Depletion and Remissions of Malignancy along with Cytokine-Associated Toxicity in a Clinical Trial of Anti-CD19 Chimeric-Antigen-Receptor-Transduced T Cells. Blood 2012, 119, 2709–2720.
- Canna, S.W.; Marsh, R.A. Pediatric Hemophagocytic Lymphohistiocytosis. Blood 2020, 135, 1332–1343.
- Cardiotoxicity from Chimeric Antigen Receptor-T Cell Therapy for Advanced Malignancies. Available online: https://pubmed.ncbi.nlm.nih.gov/35257157/ (accessed on 3 April 2023).
- Camilli, M.; Maggio, L.; Tinti, L.; Lamendola, P.; Lanza, G.A.; Crea, F.; Lombardo, A. Chimeric Antigen Receptor-T Cell Therapy-Related Cardiotoxicity in Adults and Children Cancer Patients: A Clinical Appraisal. Front. Cardiovasc. Med. 2023, 10, 1090103.
- Duan, D.; Wang, K.; Wei, C.; Feng, D.; Liu, Y.; He, Q.; Xu, X.; Wang, C.; Zhao, S.; Lv, L.; et al. The BCMA-Targeted Fourth-Generation CAR-T Cells Secreting IL-7 and CCL19 for Therapy of Refractory/Recurrent Multiple Myeloma. Front. Immunol. 2021, 12, 609421.
- Lee, D.W.; Santomasso, B.D.; Locke, F.L.; Ghobadi, A.; Turtle, C.J.; Brudno, J.N.; Maus, M.V.; Park, J.H.; Mead, E.; Pavletic, S.; et al. ASTCT Consensus Grading for Cytokine Release Syndrome and Neurologic Toxicity Associated with Immune Effector Cells. Biol. Blood Marrow Transplant. 2019, 25, 625–638.
- Gust, J.; Hay, K.A.; Hanafi, L.-A.; Li, D.; Myerson, D.; Gonzalez-Cuyar, L.F.; Yeung, C.; Liles, W.C.; Wurfel, M.; Lopez, J.A.; et al. Endothelial Activation and Blood-Brain Barrier Disruption in Neurotoxicity after Adoptive Immunotherapy with CD19 CAR-T Cells. Cancer Discov. 2017, 7, 1404–1419.
- Giavridis, T.; van der Stegen, S.J.C.; Eyquem, J.; Hamieh, M.; Piersigilli, A.; Sadelain, M. CAR T Cell-Induced Cytokine Release Syndrome Is Mediated by Macrophages and Abated by IL-1 Blockade. Nat. Med. 2018, 24, 731–738.
- Norelli, M.; Camisa, B.; Barbiera, G.; Falcone, L.; Purevdorj, A.; Genua, M.; Sanvito, F.; Ponzoni, M.; Doglioni, C.; Cristofori, P.; et al. Monocyte-Derived IL-1 and IL-6 Are Differentially Required for Cytokine-Release Syndrome and Neurotoxicity Due to CAR T Cells. Nat. Med. 2018, 24, 739–748.
- Predominant Cerebral Cytokine Release Syndrome in CD19-Directed Chimeric Antigen Receptor-Modified T Cell Therapy. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4986179/ (accessed on 6 April 2023).
- Parker, K.R.; Migliorini, D.; Perkey, E.; Yost, K.E.; Bhaduri, A.; Bagga, P.; Haris, M.; Wilson, N.E.; Liu, F.; Gabunia, K.; et al. Single-Cell Analyses Identify Brain Mural Cells Expressing CD19 as Potential Off-Tumor Targets for CAR-T Immunotherapies. Cell 2020, 183, 126–142.e17.
- Neelapu, S.S.; Tummala, S.; Kebriaei, P.; Wierda, W.; Gutierrez, C.; Locke, F.L.; Komanduri, K.V.; Lin, Y.; Jain, N.; Daver, N.; et al. Chimeric Antigen Receptor T-Cell Therapy—Assessment and Management of Toxicities. Nat. Rev. Clin. Oncol. 2018, 15, 47–62.
- Hunter, B.D.; Jacobson, C.A. CAR T-Cell Associated Neurotoxicity: Mechanisms, Clinicopathologic Correlates, and Future Directions. J. Natl. Cancer Inst. 2019, 111, 646–654.
- Sheth, V.S.; Gauthier, J. Taming the Beast: CRS and ICANS after CAR T-Cell Therapy for ALL. Bone Marrow Transplant. 2021, 56, 552–566.
- Barbar, T.; Jaffer Sathick, I. Tumor Lysis Syndrome. Adv. Chronic Kidney Dis. 2021, 28, 438–446.e1.
- Maus, M.V.; Haas, A.R.; Beatty, G.L.; Albelda, S.M.; Levine, B.L.; Liu, X.; Zhao, Y.; Kalos, M.; June, C.H. T Cells Expressing Chimeric Antigen Receptors Can Cause Anaphylaxis in Humans. Cancer Immunol. Res. 2013, 1, 26–31.
- Kochenderfer, J.N.; Dudley, M.E.; Carpenter, R.O.; Kassim, S.H.; Rose, J.J.; Telford, W.G.; Hakim, F.T.; Halverson, D.C.; Fowler, D.H.; Hardy, N.M.; et al. Donor-Derived CD19-Targeted T Cells Cause Regression of Malignancy Persisting after Allogeneic Hematopoietic Stem Cell Transplantation. Blood 2013, 122, 4129–4139.
- Thistlethwaite, F.C.; Gilham, D.E.; Guest, R.D.; Rothwell, D.G.; Pillai, M.; Burt, D.J.; Byatte, A.J.; Kirillova, N.; Valle, J.W.; Sharma, S.K.; et al. The Clinical Efficacy of First-Generation Carcinoembryonic Antigen (CEACAM5)-Specific CAR T Cells Is Limited by Poor Persistence and Transient Pre-Conditioning-Dependent Respiratory Toxicity. Cancer Immunol. Immunother. 2017, 66, 1425–1436.
- Flugel, C.L.; Majzner, R.G.; Krenciute, G.; Dotti, G.; Riddell, S.R.; Wagner, D.L.; Abou-el-Enein, M. Overcoming On-Target, off-Tumour Toxicity of CAR T Cell Therapy for Solid Tumours. Nat. Rev. Clin. Oncol. 2022, 20, 49–62.
- Mueller, K.T.; Maude, S.L.; Porter, D.L.; Frey, N.; Wood, P.; Han, X.; Waldron, E.; Chakraborty, A.; Awasthi, R.; Levine, B.L.; et al. Cellular Kinetics of CTL019 in Relapsed/Refractory B-Cell Acute Lymphoblastic Leukemia and Chronic Lymphocytic Leukemia. Blood 2017, 130, 2317–2325.
- Fajgenbaum, D.C.; June, C.H. Cytokine Storm. N. Engl. J. Med. 2020, 383, 2255–2273.
- Brentjens, R.J.; Davila, M.L.; Riviere, I.; Park, J.; Wang, X.; Cowell, L.G.; Bartido, S.; Stefanski, J.; Taylor, C.; Olszewska, M.; et al. CD19-Targeted T Cells Rapidly Induce Molecular Remissions in Adults with Chemotherapy-Refractory Acute Lymphoblastic Leukemia. Sci. Transl. Med. 2013, 5, 177ra38.
- Finney, O.C.; Brakke, H.M.; Rawlings-Rhea, S.; Hicks, R.; Doolittle, D.; Lopez, M.; Futrell, R.B.; Orentas, R.J.; Li, D.; Gardner, R.A.; et al. CD19 CAR T Cell Product and Disease Attributes Predict Leukemia Remission Durability. J. Clin. Investig. 2019, 129, 2123–2132.
- Davila, M.L.; Riviere, I.; Wang, X.; Bartido, S.; Park, J.; Curran, K.; Chung, S.S.; Stefanski, J.; Borquez-Ojeda, O.; Olszewska, M.; et al. Efficacy and Toxicity Management of 19-28z CAR T Cell Therapy in B Cell Acute Lymphoblastic Leukemia. Sci. Transl. Med. 2014, 6, 224ra25.
- Zhang, A.; Sun, Y.; Du, J.; Dong, Y.; Pang, H.; Ma, L.; Si, S.; Zhang, Z.; He, M.; Yue, Y.; et al. Reducing Hinge Flexibility of CAR-T Cells Prolongs Survival In Vivo With Low Cytokines Release. Front. Immunol. 2021, 12, 724211.
- Ying, Z.; Huang, X.F.; Xiang, X.; Liu, Y.; Kang, X.; Song, Y.; Guo, X.; Liu, H.; Ding, N.; Zhang, T.; et al. A Safe and Potent Anti-CD19 CAR T Cell Therapy. Nat. Med. 2019, 25, 947–953.
- Alabanza, L.; Pegues, M.; Geldres, C.; Shi, V.; Wiltzius, J.J.W.; Sievers, S.A.; Yang, S.; Kochenderfer, J.N. Function of Novel Anti-CD19 Chimeric Antigen Receptors with Human Variable Regions Is Affected by Hinge and Transmembrane Domains. Mol. Ther. 2017, 25, 2452–2465.
- Pietrobon, V.; Todd, L.A.; Goswami, A.; Stefanson, O.; Yang, Z.; Marincola, F. Improving CAR T-Cell Persistence. Int. J. Mol. Sci. 2021, 22, 10828.
- Brudno, J.N.; Maric, I.; Hartman, S.D.; Rose, J.J.; Wang, M.; Lam, N.; Stetler-Stevenson, M.; Salem, D.; Yuan, C.; Pavletic, S.; et al. T Cells Genetically Modified to Express an Anti-B-Cell Maturation Antigen Chimeric Antigen Receptor Cause Remissions of Poor-Prognosis Relapsed Multiple Myeloma. J. Clin. Oncol. 2018, 36, 2267–2280.