Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Camila Xu and Version 1 by Dimitar Georgiev Tonev.
Multiple sclerosis (MS) is predominantly an immune-mediated disease of the central nervous system (CNS) of unknown etiology with a possible genetic predisposition and effect of certain environmental factors. It is generally accepted that the disease begins with an autoimmune inflammatory reaction targeting oligodendrocytes followed by a rapid depletion of their regenerative capacity with subsequent permanent neurodegenerative changes and disability.
- multiple sclerosis
- immune pathophysiology
- autoantibodies
- nerve growth factor
1. Introduction
Multiple sclerosis (MS) is an autoimmune multifocal central nervous system (CNS) inflammatory disease, characterized by chronic inflammation, demyelination, axonal damage, and subsequent gliosis. The disease is of unknown etiology with a possible genetic predisposition and effect of certain environmental factors [1,2][1][2]. Although there is no complete consensus regarding the pathological processes leading to MS, it is generally accepted that the disease begins with an autoimmune inflammatory reaction targeting oligodendrocytes (OLs) followed by a rapid depletion of OLs regenerative capacity with subsequent permanent neurodegenerative changes and disability [3]. Therefore, the therapeutic efforts should be focused with priority on the first stage of demyelination, when the damage is not yet irreversible. The most efficient treatment modality for neurodegeneration remains an early and aggressive anti-inflammatory intervention, because the prevention of tissue injury may best control the escalating T-cell-driven and bystander B-cell activation, ongoing breakdown of blood–brain barrier (BBB) and epitope spreading, that may perpetuate neuroaxonal damage [4]. After the impressive efficacy of anti-CD20 antibody therapy for patients with a relapsing–remitting form of the disease, there is renewed attention to B cells in the pathogenesis of MS [5]. Recent research highlights the central role of B lymphocytes in the development of MS lesions, in particular the main role of IgG and IgM in newly forming lesions [6]. Thus, early and aggressive control of antibodies contributing to oligodendrocyte and axonal damage in MS becomes of utmost importance. However, the questionable efficacy of anti-CD20 therapy in reducing the antibody levels [7], along with its delayed onset of action compared to the rapid action of therapeutic apheresis, raised issues of combination therapies in general [8] and between apheresis and anti-CD20 antibody therapy in particular [9]. On the other hand, although pathogenetically justified, the role of apheresis (or therapeutic plasma exchange (TPE), a term used interchangeably) in MS patients still has a limited and even questioned application [7,10][7][10]. As usually happens, the latter critical review could pave the way for searching for new answers. It was suggested that further studies should be undertaken to determine the levels and precise effects of both beneficial and harmful components in sera of MS patients during different phases of the disease in terms of their capability to serve as markers for appropriate TPE protocols [10].
2. Pathogenesis of MS
2.1. Between Space and Time Axes
MS is an autoimmune multifocal CNS inflammatory disease, characterized by chronic inflammation, demyelination, axonal damage, and subsequent gliosis. The most affected CNS regions by the disease are the periventricular area, subcortical area, optic nerve, spinal cord, brainstem, and cerebellum. MS is categorized as relapsing–remitting (RR), secondary progressive (SP), and primary progressive (PP) [11]. The pathogenesis of MS suggests that in genetically susceptible subjects, independent populations of T lymphocytes are activated in the immune system, migrate across the BBB, and trigger CNS tissue damage. They release pro-inflammatory cytokines, initiate cytotoxic activities of microglia with the release of nitrous oxide and other superoxide radicals, stimulate B cells and macrophages, and activate the complement system [12]. Autoantibodies against myelin basic protein and myelin OLs glycoprotein have been detected in MS patients. These antibodies may mediate injury by complement fixation or linking with innate effector cells such as CNS resident macrophages [12]. Despite the specific clinical form, the initial stage of the disease is characterized by an autoimmune inflammatory response mainly against the OLs in the CNS, resulting in demyelination (inflammatory component). Soon after the regenerative capacity of the OLs is exhausted, the inflammatory processes attack the neurons themselves, leading to the permanent injury and dysfunction of the CNS (neurodegenerative component). Both inflammatory and neurodegenerative components of MS pathogenesis are believed to be involved from the very beginning of the disease, giving different clinical presentations in the context of spatial and temporal pathological changes) (Figure 1) [13].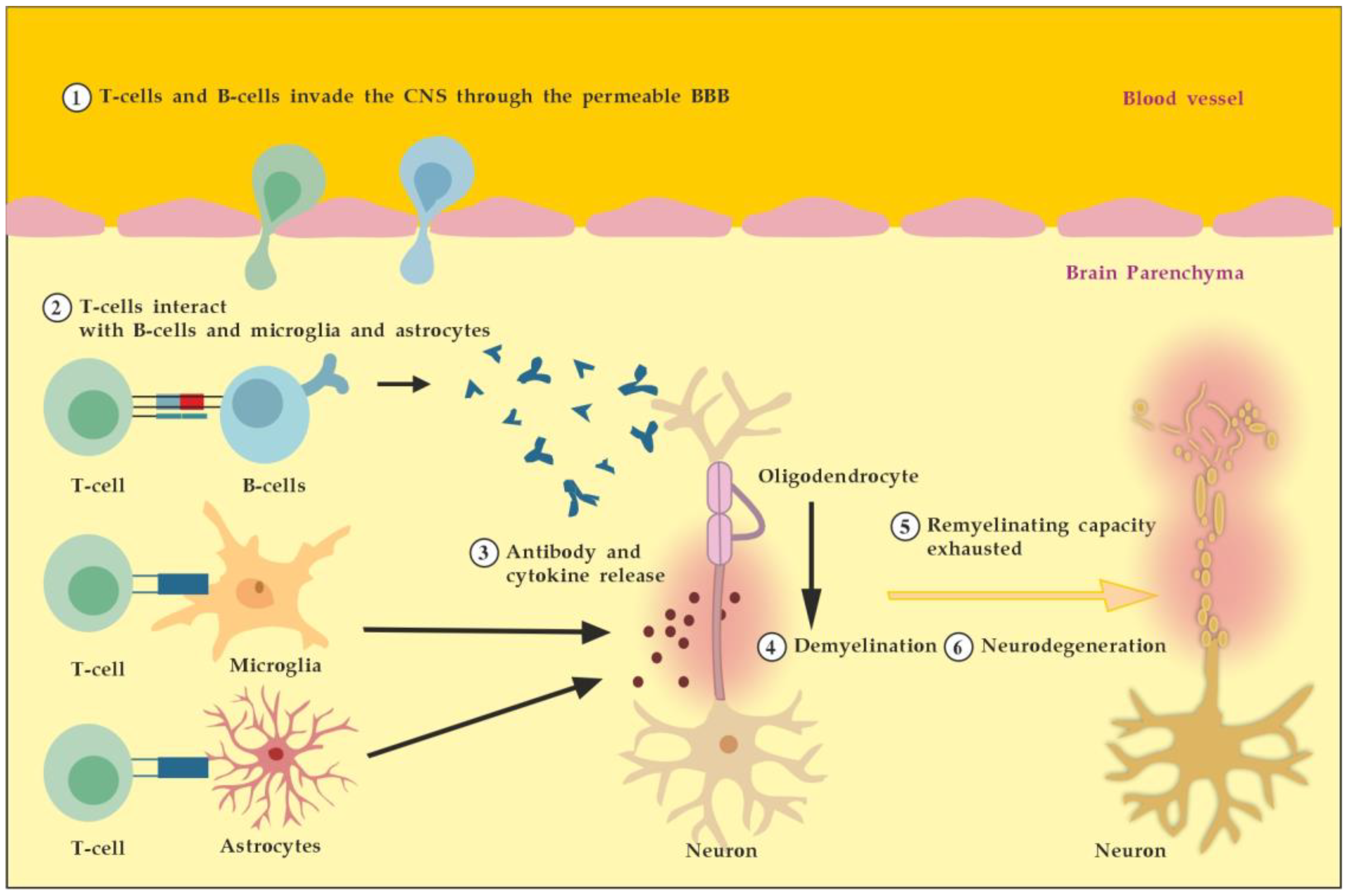
Figure 1. Pathogenesis of MS. Described processes 1 to 6 occur consecutively and in parallel in terms of the spatial (peripheral vs. CNS inflammation) and temporal (acute vs. chronic inflammation) axes. Adapted from [3] and modified.
2.2. Between Detrimental and Beneficial Neuro-Inflammatory Responses
2.2.1. The Role of Peripheral Immune Cells
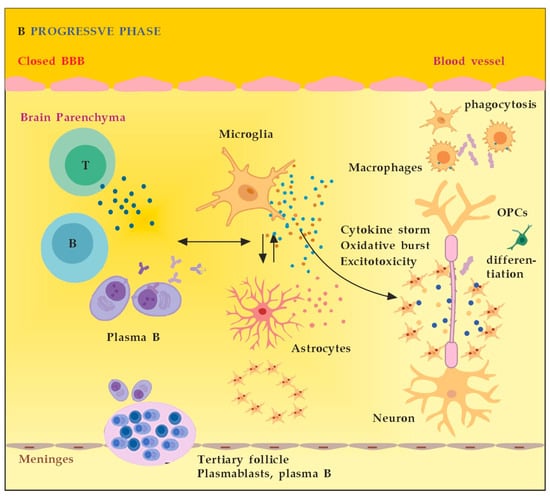
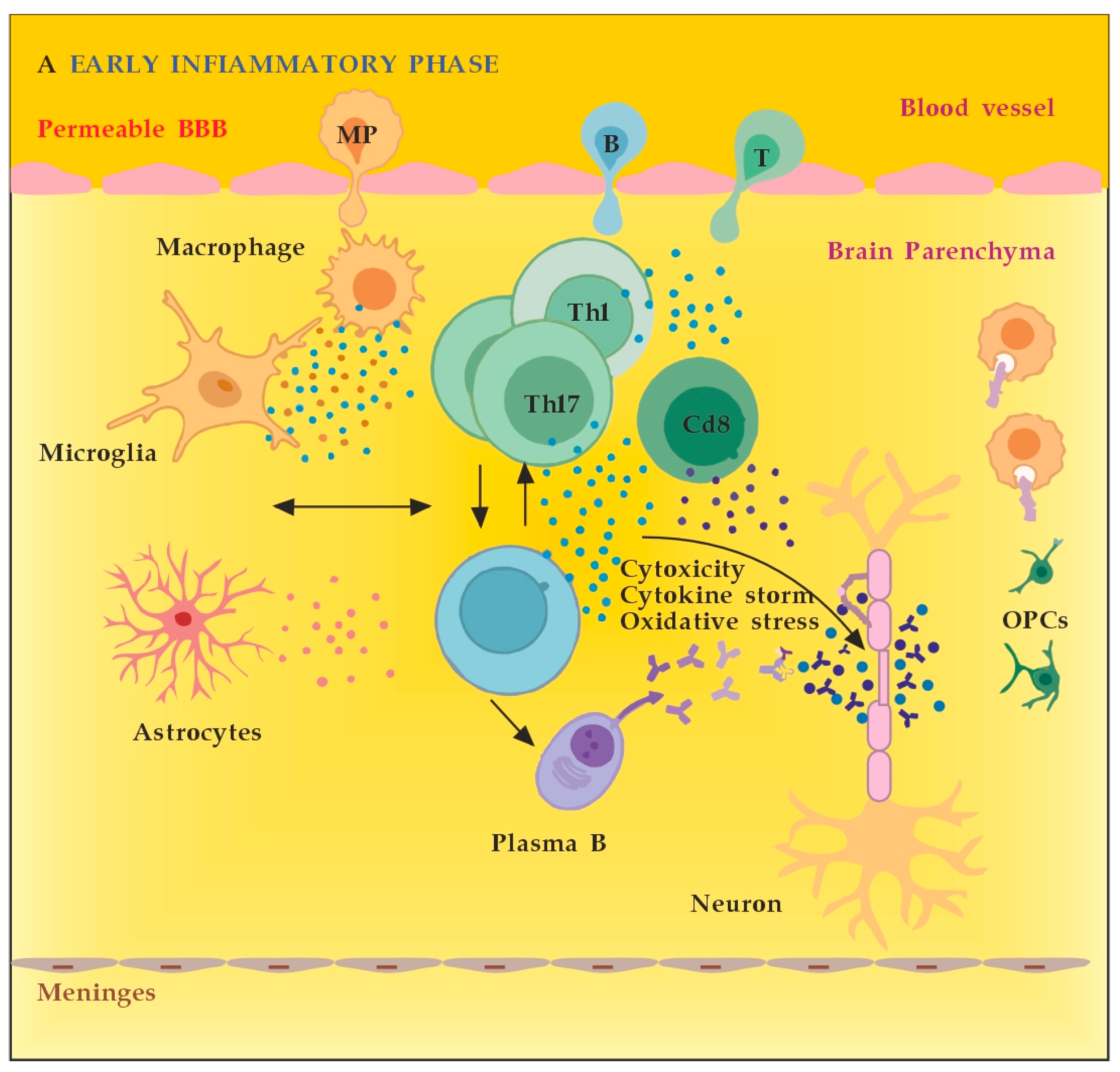
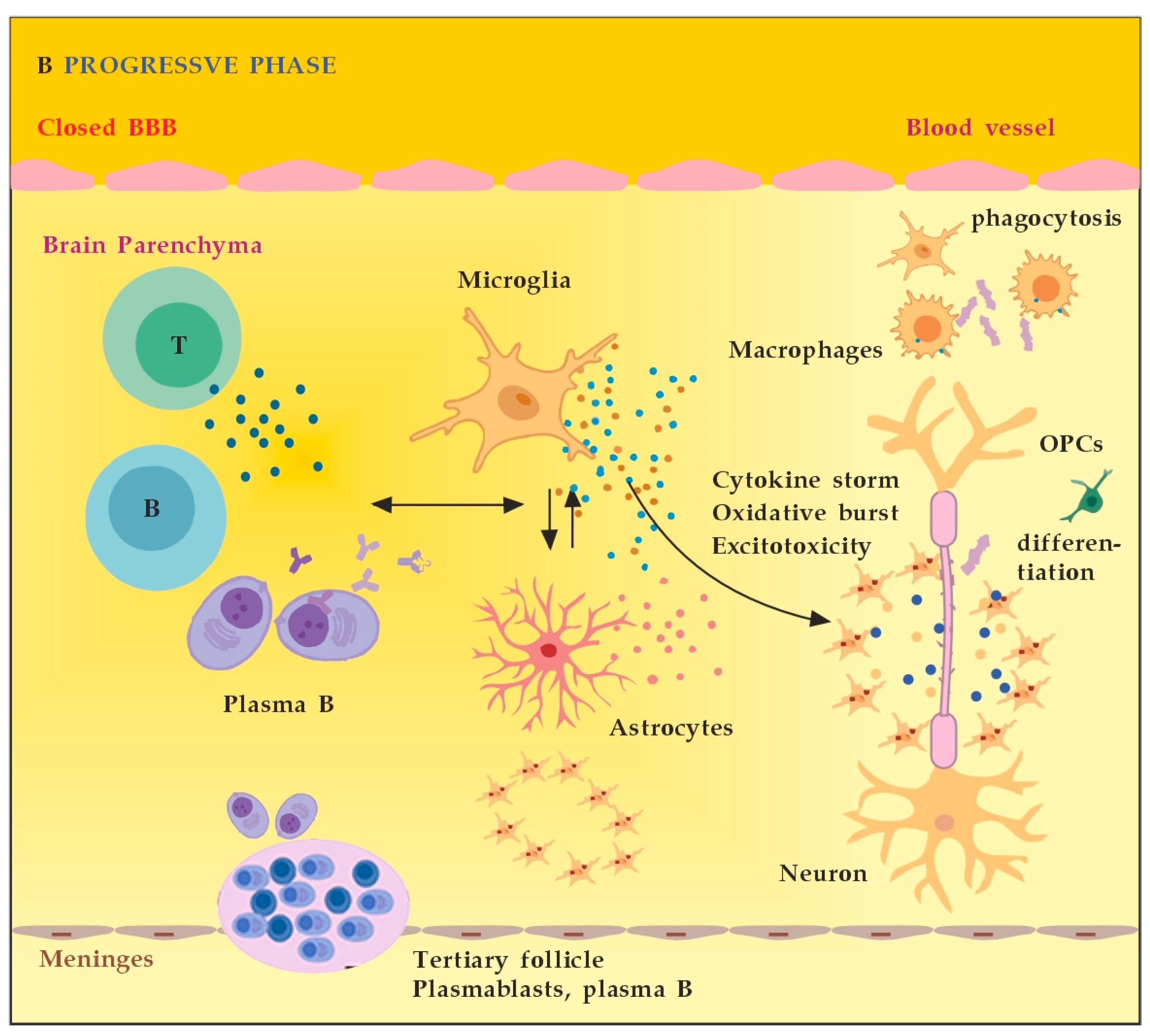
MS neuro-inflammation is characterized by pathogenic immune responses involving T cells (CD4+ and CD8+ T cells), B cells, and myeloid cells along with the reduced function of regulatory T cells [14]. In the early inflammatory phase of MS, peripheral adaptive immune cells infiltrate the CNS through a compromised BBB (Figure 2A). These activated cells interact with each other and with CNS resident cells. They secrete cytokines such as IFN-γ by Th1, IL-6, IL-17 by Th17, GM-CSF, IL-6, TNF-α by B cells, and cytotoxic molecules such as granzyme B by CD8+ T cells. B cells can further evolve into pathogenic autoantibody-producing plasma cells. As a result, T and B cells activate macrophages and microglia that produce cytokines, nitric oxide, and reactive oxygen species (ROS). This cytotoxic pro-inflammatory environment causes oligodendrocyte and axonal damage through direct cell contact-dependent processes and the release of neurotoxic mediators [13]. It destroys the myelin sheaths around axons and causes energy failure in the axon. Yet, macrophages and microglia can still clear the myelin debris, allowing for the recruitment of oligodendrocyte progenitor cells (OPCs) that will partially remyelinate the lesion [13,15][13][15]. In the progressive phase of MS, the inflammation is restricted within the CNS due to the persistence of activated immune cells in situ, despite the absence of infiltrating T and B lymphocytes from the periphery (Figure 2B). This chronic inflammatory process affects the whole brain parenchyma, even at sites distant from the underlying focal demyelinating lesions. Diffuse chronic CNS inflammation is thought to be more common in patients with progressive forms of MS [16,17][16][17]. Notably, plasmablasts and plasma B cells form tertiary follicle-like structures in the meninges. Their location in the leptomeninges contributes to the demyelination of subpial gray matter and highlights the importance of B lymphocytes in the pathogenesis of the progressive form of MS. The BBB is closed and the inflammation is maintained by innate resident CNS cells, i.e., microglia and astrocytes. They produce cytokines (TNF-α, IL-6) and release ROS, causing damage to myelin [18]. Recent data suggest that neuro-inflammation may be beneficial to some extent [19]. In experimental autoimmune encephalomyelitis (EAE) models, the treatment of mice with IFN-γ, classically considered a pro-inflammatory cytokine, leads to reduced morbidity and mortality [20]. Evidence also supports the protective role of tumor necrosis factor alpha (TNF-α) in EAE. Mice lacking TNF-α and its related receptors showed a significant delay in remyelination [21]. This could be related to the missing TNF-α induction of neurotrophins expression as well, such as NGF and brain-derived neurotrophic factor (BDNF) [22]. TNF-α treatment significantly reduced the severity of the disease in immunized TNF-deficient mice [23]. The results suggest that some pro-inflammatory cytokines may also play an indirect rather than direct role in disease control and remyelination [20,21,23][20][21][23]. Anti-inflammatory cytokines, such as IL-4 and IL-10, may have a direct protective effect instead [24,25][24][25]. Immune cells also exert a neuroprotective effect in MS via the production and local secretion of neurotrophins, such as NGF and BDNF [26]. In addition, after suppressing B cell cytokines BAFF and APRIL with atacicept (cytokines important for B cell survival and function), adversely increased clinical activity in MS was observed. The latter provides indirect evidence for the anti-inflammatory functions of certain B cells [27].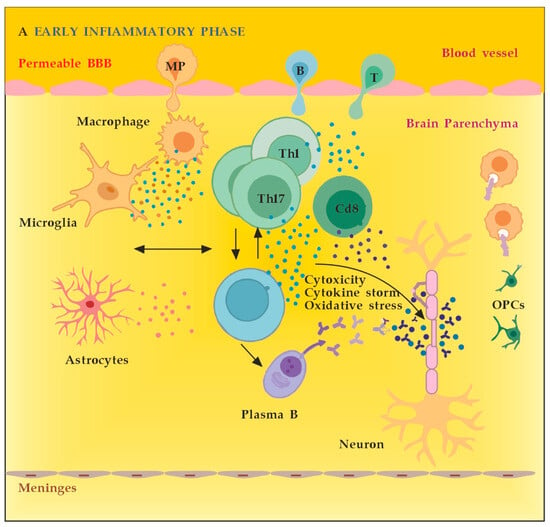
2.2.2. The Role of the Innate Resident CNS Cells
The main innate resident CNS cells of relevance to the localized inflammatory response are microglia and astrocytes. They produce cytokines (TNF-α, IL-6) and release ROS in situ, causing myelin damage [28]. Microglia are activated by pathogen-associated molecular pattern molecules (PAMPs) and/or damage-associated molecular pattern molecules (DAMPs) [29]. The classical (M1) microglia activation produces pro-inflammatory cytokines and chemokines, such as tumor necrosis factor-alpha (TNF-α), interleukin (IL)-6, IL-1β, IL-12, and CC chemokine ligand 2 (CCL2), and induce inflammation and neurotoxicity) [30]. The alternative (M2) microglia activation secretes certain growth factors (GFs) and neurotrophic factors (NTFs) and promotes the survival of neurons [31]. The switch from M1 to M2 phenotype may occur via inhibition of nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), mitogen-activated protein kinase (MAPK), activator protein 1 (AP-1), and signal transducer and activator of transcription (STAT) transcription factors, and activation of the peroxisome proliferator–activated receptor gamma (PPARγ) pathway [31,32,33,34,35,36][31][32][33][34][35][36] (Figure 3). Microglia activation contributes to MS disregulation through antigen presentation, secretion of pro-inflammatory cytokines, and phagocytic processes [37,38][37][38]. Although microglia are primed into a pro-inflammatory phenotype, their phagocytic capacities are diminished. Myelin debris are not completely cleared, OPCs are insufficiently recruited and fail to differentiate. Overall, microglial activation in the CNS is heterogeneous and cannot be classified only into two different subtypes: classical (M1) or alternative (M2) [39]. Although M1 microglia promote inflammation and M2 microglia have an anti-inflammatory phenotype, there appears to be a continuum of phenotypes between M1 and M2 that can switch from one to another [40]. Modulation microglia M1/M2 polarization and shifting from M1 to M2 phenotype have been proposed as promising therapeutic strategies in neurodegenerative CNS disorders [41]. Astrocytes may play a role in inhibiting remyelination and axonal regeneration through reactive astrogliosis, glial scar formation, and the secretion of inhibitory molecules which suppress axonal growth [42]. TNF-α-mediated glutamate release from astrocytes leads to excitotoxicity, causing axonal damage. The ferrous iron (Fe2+) released from the myelin is oxidized to produce ROS leading to a major oxidative burst, causing mitochondrial dysfunction, mitochondrial DNA damage, energy failure, and axonal loss [18]. Activated astrocytes show a Janus-faced nature. The A1 astrocytes secrete interleukin (IL)-1β, TNF-α, and C3 components to propagate the neuroinflammatory response. Additionally, they also secrete D-serine and nitric oxide (NO), which may contribute to excitotoxicity with subsequent neuronal and oligodendrocyte death. The alternative A2 astrocytes, however, may secrete anti-inflammatory compounds, such as neurotrophic factors (NTFs, including NGF), IL-10, IL-6, and TGF-β, and promote the neuroprotective and neuroregenerative functions [41,43][41][43]. Astrocytes may stop the T-cell response by inducing apoptosis as well [44]. Until recently, astrocytes’ formation of the glial scar was considered a harmful process that impedes the regeneration and remyelination of axons. Seen from a different perspective, however, depending on the severity of the injury, the scarring process may also serve to isolate the inflamed area, provide structural support, and restrict damage [42]. Likewise, activated microglia can also promote remyelination by clearing myelin debris from the local environment and by secreting anti-inflammatory cytokines, such as transforming growth factor-beta 1 (TGF-ß1) and certain neurotrophic factors, such as BDNF, that can induce the proliferation of OLs (Figure 3) [45,46][45][46].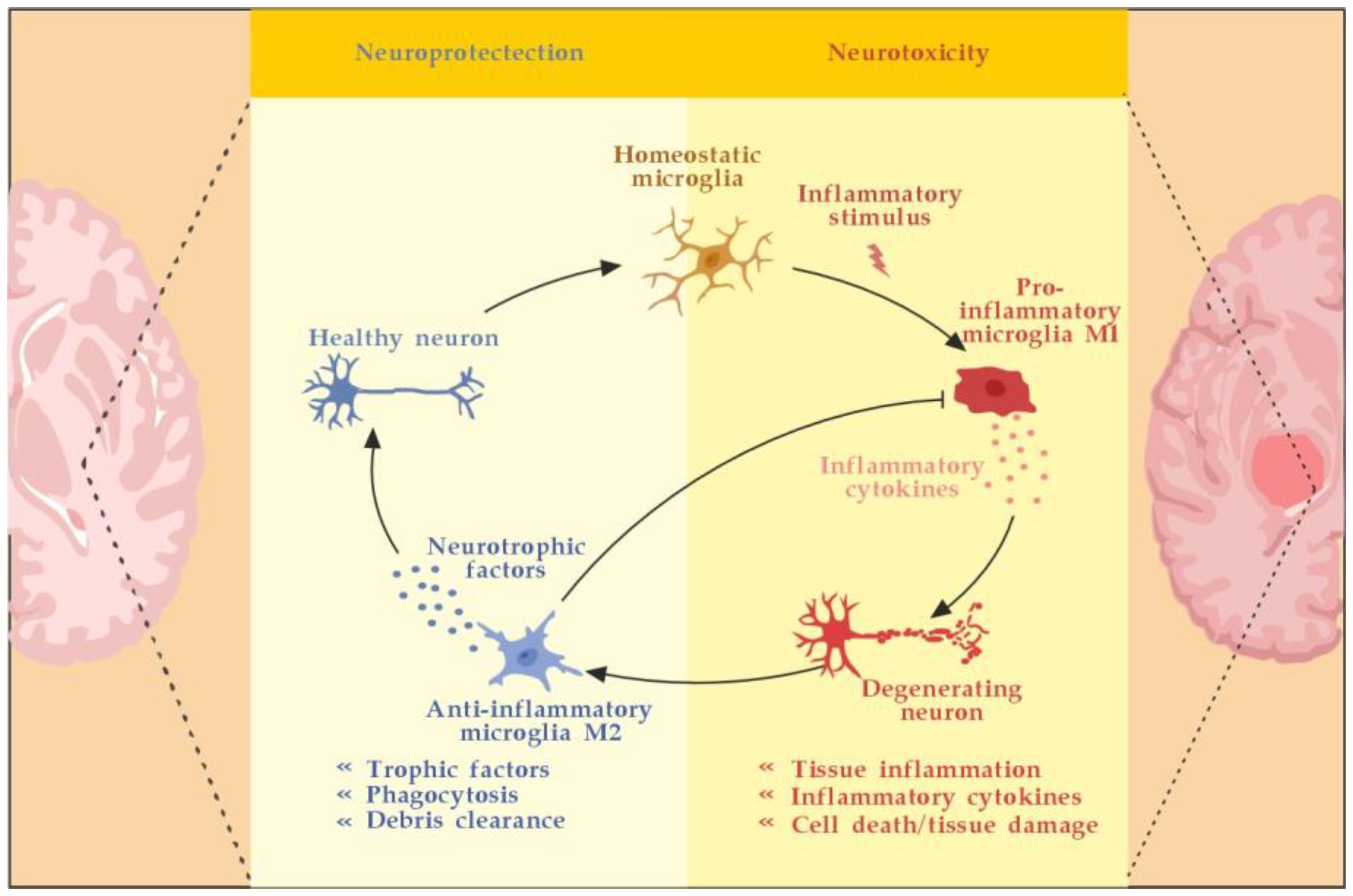
References
- Zahoor, I.; Rui, B.; Khan, J.; Datta, I.; Giri, S. An emerging potential of metabolomics in multiple sclerosis: A comprehensive overview. Cell. Mol. Life Sci. 2021, 78, 3181–3203.
- Lassmann, H. Pathogenic mechanisms associated with different clinical courses of multiple sclerosis. Front. Immunol. 2018, 9, 3116.
- Sandi, D.; Kokas, Z.; Biernacki, T.; Bencsik, K.; Klivényi, P.; Vécsei, L. Proteomics in Multiple Sclerosis: The Perspective of the Clinician. Int. J. Mol. Sci. 2022, 23, 5162.
- Vyshkina, T.; Kalman, B. Autoantibodies and neurodegeneration in multiple sclerosis. Lab. Investig. 2008, 88, 796–807.
- Li, Y.; Noto, D.; Hoshino, Y.; Mizuno, M.; Yoshikawa, S.; Miyake, S. Immunoglobulin directly enhances differentiation of oli-godendrocyte-precursor cells and remyelination. Sci. Rep. 2023, 13, 9394.
- Muñoz, U.; Sebal, C.; Escudero, E.; Esiri, M.; Tzartos, J.; Sloan, C.; Sadaba, M.C. Main Role of Antibodies in Demyelination and Axonal Damage in Multiple Sclerosis. Cell. Mol. Neurobiol. 2022, 42, 1809–1827.
- Höftberger, R.; Lassmann, H.; Berger, T.; Reindl, M. Pathogenic autoantibodies in multiple sclerosis—From a simple idea to a complex concept. Nat. Rev. Neurol. 2022, 18, 681–688.
- Cunniffe, N.; Coles, A. Promoting remyelination in multiple sclerosis. J. Neurol. 2021, 268, 30–44.
- Davies, A.J.; Fehmi, J.; Senel, M.; Tumani, H.; Dorst, J.; Rinaldi, S. Immunoadsorption and Plasma Exchange in Seropositive and Seronegative Immune-Mediated Neuropathies. J. Clin. Med. 2020, 9, 2025.
- Farrokhi, M. Plasmapheresis for Multiple Sclerosis in the Twenty-First Century: Take It or Leave It? J. Rev. Med. Sci. 2021, 1, e1.
- Lublin, F.D.; Reingold, S.C.; Cohen, J.A.; Cutter, G.R.; Sørensen, P.S.; Thompson, A.J.; Wolinsky, J.S.; Balcer, L.J.; Banwell, B.; Barkhof, F.; et al. Defining the clinical course of multiple sclerosis: The 2013 revisions. Neurology 2014, 83, 278–286.
- Lassmann, H.; Brück, W.; Lucchinetti, C.F. The Immunopathology of Multiple Sclerosis: An Overview. Brain Pathol. 2007, 17, 210–218.
- Dendrou, C.A.; Fugger, L.; Friese, M.A. Immunopathology of multiple sclerosis. Nat. Rev. Immunol. 2015, 15, 545–558.
- Danikowski, K.M.; Jayaraman, S.; Prabhakar, B.S. Regulatory T cells in multiple sclerosis and myasthenia gravis. J. Neuroinflammation 2017, 14, 117.
- Haase, S.; Linker, R.A. Inflammation in multiple sclerosis. Ther. Adv. Neurol. Disord. 2021, 14, 17562864211007687.
- Bordet, R.; Camu, W.; De Seze, J.; Laplaud, D.-A.; Ouallet, J.-C.; Thouvenot, E. Mechanism of action of s1p receptor modulators in multiple sclerosis: The double requirement. Rev. Neurol. 2020, 176, 100–112.
- Stadelmann, C.; Wegner, C.; Bruck, W. Inflammation, demyelination, and degeneration—Recent insights from MS pathology. Biochim. Biophys. Acta 2011, 1812, 275–282.
- Perdaens, O.; van Pesch, V. Molecular Mechanisms of Immunosenescene and Inflammaging: Relevance to the Immunopath-ogenesis and Treatment of Multiple Sclerosis. Front. Neurol. 2022, 12, 811518.
- Martino, G.; Adorini, L.; Rieckmann, P.; Hillert, J.; Kallmann, B.; Comi, G.; Filippi, M. Inflammation in multiple sclerosis: The good, the bad, and the complex. Lancet Neurol. 2002, 1, 499–509.
- Billiau, A.; Heremans, H.; Vandekerckhove, F.; Dijkmans, R.; Sobis, H.; Meulepas, E.; Carton, H. Enhancement of experimental allergic encephalomyelitis in mice by antibodies against IFN-gamma. J. Immunol. 1988, 140, 1506–1510.
- Arnett, H.A.; Mason, J.; Marino, M.; Suzuki, K.; Matsushima, G.K.; Ting, J.P. TNF alpha promotes proliferation of oligoden-drocyte progenitors and remyelination. Nat. Neurosci. 2001, 4, 1116–1122.
- Acosta, M.; Cortes, C.; MacPhee, H.; Namaka, M.P. Exploring the role of nerve growth factor in multiple sclerosis: Implications in myelin repair. CNS Neurol. Disord.—Drug Targets 2013, 12, 1242–1256.
- Liu, J.; Marino, M.W.; Wong, G.; Grail, D.; Dunn, A.; Bettadapura, J.; Slavin, A.J.; Old, L.; Bernard, C.C. TNF is a potent an-ti-inflammatory cytokine in autoimmune-mediated demyelination. Nat. Med. 1998, 4, 78–83.
- Wang, K.; Song, F.; Fernandez-Escobar, A.; Luo, G.; Wang, J.-H.; Sun, Y. The Properties of Cytokines in Multiple Sclerosis: Pros and Cons. Am. J. Med. Sci. 2018, 356, 552–560.
- Hulshof, S.; Montagne, L.; De Groot, C.J.; Van Der Valk, P. Cellular localization and expression patterns of interleukin-10, interleukin-4, and their receptors in multiple sclerosis lesions. Glia 2002, 38, 24–35.
- Kerschensteiner, M.; Gallmeier, E.; Behrens, L.; Leal, V.V.; Misgeld, T.; Klinkert, W.E.; Kolbeck, R.; Hoppe, E.; Oropeza-Wekerle, R.L.; Bartke, I.; et al. Activated human T cells, B cells, and monocytes produce brain-derived neurotrophic factor in vitro and in inflammatory brain lesions: A neuroprotective role of inflammation? J. Exp. Med. 1999, 189, 865–870.
- Kappos, L.; Hartung, H.-P.; Freedman, M.S.; Boyko, A.; Radü, E.W.; Mikol, D.D.; Lamarine, M.; Hyvert, Y.; Freudensprung, U.; Plitz, T.; et al. Atacicept in multiple sclerosis (ATAMS): A randomised, placebo-controlled, double-blind, phase 2 trial. Lancet Neurol. 2014, 13, 353–363.
- Nociti, V.; Romozzi, M. The Role of BDNF in Multiple Sclerosis Neuroinflammation. Int. J. Mol. Sci. 2023, 24, 8447.
- Rodríguez, A.M.; Rodríguez, J.; Giambartolomei, G.H. Microglia at the Crossroads of Pathogen-Induced Neuroinflammation. ASN Neuro 2022, 14, 175909142211045.
- Colonna, M.; Butovsky, O. Microglia Function in the Central Nervous System During Health and Neurodegeneration. Annu. Rev. Immunol. 2017, 35, 441–468.
- Guo, S.; Wang, H.; Yin, Y. Microglia Polarization From M1 to M2 in Neurodegenerative Diseases. Front. Aging Neurosci. 2022, 14, 815347.
- Liu, X.; Ma, J.; Ding, G.; Gong, Q.; Wang, Y.; Yu, H.; Cheng, X. Microglia Polarization from M1 toward M2 Phenotype Is Promoted by Astragalus Polysaccharides Mediated through Inhibition of MiR-155 in Experimental Autoimmune Encephalo-myelitis. Oxidative Med. Cell. Longev. 2021, 2021, 5753452.
- Qie, S.; Ran, Y.; Lu, X.; Su, W.; Li, W.; Xi, J.; Gong, W.; Liu, Z. Candesartan Modulates Microglia Activation and Polarization via NF-KB Signaling Pathway. Int. J. Immunopathol. Pharmacol. 2020, 34, 205873842097490.
- Zhang, B.; Wei, Y.-Z.; Wang, G.-Q.; Li, D.-D.; Shi, J.-S.; Zhang, F. Targeting MAPK Pathways by Naringenin Modulates Mi-croglia M1/M2 Polarization in Lipopolysaccharide-Stimulated Cultures. Front. Cell Neurosci. 2019, 12, 531.
- Ding, Y.; Kang, J.; Liu, S.; Xu, Y.; Shao, B. The Protective Effects of Peroxisome Proliferator-Activated Receptor Gamma in Cerebral Ischemia-Reperfusion Injury. Front. Neurol. 2020, 11, 588516.
- Guerrero, B.L.; Sicotte, N.L. Microglia in Multiple Sclerosis: Friend or Foe? Front. Immunol. 2020, 11, 374.
- Gandhi, R.; Laroni, A.; Weiner, H.L. Role of the innate immune system in the pathogenesis of multiple sclerosis. J. Neuroimmunol. 2010, 221, 7–14.
- Luo, C.; Jian, C.; Liao, Y.; Huang, Q.; Wu, Y.; Liu, X.; Zou, D.; Wu, Y. The role of microglia in multiple sclerosis. Neuropsychiatr. Dis. Treat. 2017, 13, 1661–1667.
- Tang, Y.; Le, W. Differential Roles of M1 and M2 Microglia in Neurodegenerative Diseases. Mol. Neurobiol. 2016, 53, 1181–1194.
- Cherry, J.D.; Olschowka, J.A.; O’Banion, M.K. Neuroinflammation and M2 microglia: The good, the bad, and the inflamed. J. Neuroinflamm. 2014, 11, 98.
- Palasz, E.; Wilkaniec, A.; Stanaszek, L.; Andrzejewska, A.; Adamczyk, A. Glia-Neurotrophic Factor Relationships: Possible Role in Pathobiology of Neuroinflammation-Related Brain Disorders. Int. J. Mol. Sci. 2023, 24, 6321.
- Sofroniew, M.V.; Vinters, H.V. Astrocytes: Biology and pathology. Acta Neuropathol. 2010, 119, 7–35.
- Ponath, G.; Park, C.; Pitt, D. The Role of Astrocytes in Multiple Sclerosis. Front. Immunol. 2018, 9, 217.
- Correale, J.; Farez, M.F. The Role of Astrocytes in Multiple Sclerosis Progression. Front. Neurol. 2015, 6, 180.
- Voss, E.V.; Škuljec, J.; Gudi, V.; Skripuletz, T.; Pul, R.; Trebst, C.; Stangel, M. Characterisation of microglia during de- and remyelination: Can they create a repair promoting environment? Neurobiol. Dis. 2012, 45, 519–528.
- Hao, Z.; Liu, K.; Zhou, L.; Chen, P. Precious but convenient means of prevention and treatment: Physiological molecular mechanisms of interaction between exercise and motor factors and Alzheimer’s disease. Front. Physiol. 2023, 14, 1193031.
More