The severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), the etiological culprit of COronaVIrus Disease 19 (COVID-19), can enter the cells via the angiotensin-converting enzyme 2 (ACE2) receptor, which has been found in several tissues including in endocrine organs, such as the ovaries, testes, pancreas, and thyroid. Several thyroid disorders have been associated with SARS-CoV-2 infection [subacute thyroiditis (SAT), thyrotoxicosis, and non-thyroidal illness syndrome (NTIS)] and, in part, they are believed to be secondary to the local virus replication within the gland cells. However, as documented for other viruses, also SARS-CoV-2 seems to interfere with several aspects of the immune system, inducing the synthesis of autoantibodies and triggering latent or new onset autoimmune disease (AID), including autoimmune thyroid disease (AITD), such as Hashimoto Thyroiditis (HT) and Graves’ disease (GD). Several mechanisms have been hypothesized to explain this induction of autoimmunity by SARS-CoV-2 infection: the immune system hyper-stimulation, the molecular mimicry between self-antigens of the host and the virus, neutrophils extracellular traps, and finally the virus induced transcriptional changes of the immune genes; nonetheless, more evidence is needed especially from large long-term cohort studies involving COVID-19 patients, to establish or reject this pathogenetic relationship.
1. The Virus Entry inside the Cells
The SARS-CoV-2 coronavirus, the etiological culprit of COVID-19, can enter the cells via the angiotensin-converting enzyme 2 (ACE2) receptor. The virus surface displays the homotrimeric spike glycoprotein, constituted by the S1 and S2 subunits, which binds to ACE2
[1][14]. During this initial interaction, the S1 subunit is disconnected with the ACE2 receptor, with the necessary help of transmembrane serine protease 2 (TMPRSS2). The resulting conformational change gives stability to the S2 subunit, allowing for membrane fusion
[2][15]. The ACE2 receptor is fundamental for SARS-CoV-2 to infect cells and, differently from other coronaviruses, it does not require further co-receptors for cellular entry, (i.e., dipeptidyl peptidase 4 or aminopeptidase N)
[3][4][16,17]. Among human beings, ACE2 mRNA can be found in several tissues, including in endocrine organs, such as the ovaries, testes, pancreas, and thyroid. TMPRSS2 mRNA is also expressed in the thyroid gland, pancreas, testes, and ovaries
[5][18]. Thus, the endocrine glands are fully exposed and vulnerable to SARS-CoV-2 infection and to subsequent dysfunctions because of COVID-19.
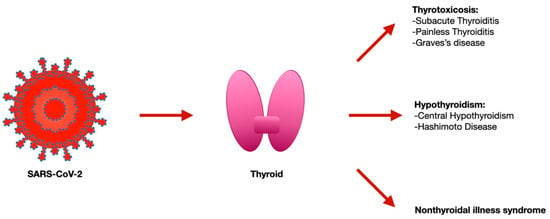
2. The Thyroid Dysfunction and SARS-CoV-2 Infection
The previous SARS pandemic already showed that affected patients had reduced thyroid function, and pathology investigations demonstrated that both thyroid follicular and parafollicular cells were extensively injured
[6][19]. Since ACE2 mRNA, together with TMPRSS2, is expressed on thyroid follicular cells
[7][20], the SARS-CoV-2 has the possibility to infect these cells as demonstrated via the autopsy report that found the viral genome and proteins inside these cells
[8][9][21,22]. Several thyroid disorders have been reported with SARS-CoV-2 infection [subacute thyroiditis (SAT), thyrotoxicosis, and non-thyroidal illness syndrome (NTIS)]
[10][11][23,24] and, in part, they are considered to be secondary to the local virus replication within the gland cells (
Figure 1).
Figure 1.
Thyroid disorders associated with SARS-CoV-2 infection.
2.1. Acute Effects
Since the beginning of the pandemic, several cases of SAT were described
[10][11][12][23,24,25]. In the ICU, patients with COVID-19 had a higher frequency of thyrotoxicosis and a lower TSH with respect to those admitted to a low-intensity ICU. However, during the 55 days of follow-up, none of them had ever complained about neck pain, but instead of lymphocytosis, they showed the typical lymphopenia of COVID-19
[13][26]. Among the COVID-19 patients not demanding intensive care management, overt thyrotoxicosis was detected in 10.8%, and hypothyroidism in 0.7%. Remarkably, thyrotoxicosis showed a correlation with serum interleukin (IL)-6 values, indicating that a greater inflammatory reaction exposed the patients to a higher chance of developing thyrotoxicosis
[11][24]. On the other hand, most of the patients (74.6%) had TSH levels within the range
[11][24], and this has been also confirmed via another sample of 334 patients with COVID-19, where 86.6% were euthyroid and none displayed overt thyrotoxicosis, even if those with COVID-19 had a lower admission of FT4 and TSH than those without COVID-19, according to an NTIS
[14][27]. NTIS, previously known as euthyroid-sick syndrome, occurs during physiological stress, especially among hospitalized patients. It consists of an initial decrease in total T3 and fT3, paralleled by an increase in reverse T3 but not of TSH
[15][28]. Long term and severe illness and ICU admission are associated with global reductions in TSH, fT4, and fT3 due to a fall in the hypothalamic thyrotropin-releasing hormone
[16][29]. These thyroid function changes in the severe illness are considered protective from excessive tissue catabolism
[17][30], and their magnitude varies with the severity of the illness: low serum T3 is associated with a longer hospital stay, ICU admission, and the need for mechanical ventilation in patients with acute heart failure
[18][31], and it predicts a 30-day mortality in patients with community-acquired pneumonia
[19][32]. For this reason, it is unsurprising that NTIS has also been detected among patients with COVID-19. The serum T4 levels also have an impact on the clinical outcomes of critically ill patients, and values lower than 3 mcg/dL have been associated with mortality rates in excess of 85 percent
[20][33]. Nevertheless, critically ill patients with low serum T3 and/or low T4 do not appear to benefit from thyroid hormone replacement therapy
[21][34]. COVID-19 pneumonia is associated with the reduced serum levels of TSH and total T3 with respect to other forms of pneumonia, but with no difference in total T4. These changes disappeared at recovery
[14][22][27,35]. In one study, among 367 patients with a complete panel of TSH, fT4, and fT4, twenty-seven patients (7.4%) had NTIS, which was associated with a higher SARS-CoV-2 viral load and inflammatory markers
[23][36]. Other studies have reported analogous data showing low TSH and/or low fT3 in patients with COVID-19
[24][25][37,38], with the amount of reduction related to the disease severity
[25][26][38,39]. Furthermore, based on the results of the RECOVERY trial
[27][40], glucocorticoids have become the standard of therapy for patients requiring oxygen supplementation and their use can lessen TSH and peripheral conversion of T4 to T3, contributing to the impairment of thyroid function, hence representing a potential confounder in the very beginning of the disease
[28][41]. Notably, thyroid diseases and their specific therapies, whether for hypothyroidism or hyperthyroidism, when controlling for relevant confounding, seem to not have an impact on the risk and prognosis of SARS-CoV-2 infection
[29][30][42,43].
2.2. Post-Acute Effects
Although COVID-19 can impair the thyroid physiology during the acute phase of the disease, patients’ statuses returned to baseline following recovery. In 68 patients that healed from COVID-19, normal thyroid function was restored in 3 to 6 months after the acute illness, displaying TSH, fT4, or fT3 levels within range
[31][44]. Moreover, Khoo and colleagues showed that 55 COVID-19 patients, whose TSH levels were also collected prior to the hospitalization (in 2019, before any cases of COVID-19), recovered to their baseline values after a median of 79 days of follow-up from the admission
[14][27]. In addition, the long-term sequalae described in patients after acute SARS-CoV-2 infection, which is known as “post-COVID syndrome” or “Long COVID”, is characterized by a protracted course of various physical and neuropsychiatric symptoms, including fatigue, anxiety, low mood, sleep disturbance, breathlessness, myalgia, and “brain fog,” which have similarities to those induced by thyroid dysfunction. Therefore, such findings will have a significant impact since they would probably increase the demand of thyroid function assessment.
2.3. Autoimmune Thyroid Diseases
More and more evidence suggests that SARS-CoV-2 is able to provoke the hyper-stimulation of the immune system, with the subsequent synthesis of several autoantibodies and the triggering of pre-existing or new onset autoimmune disease (AID), such as an antiphospholipid syndrome, autoimmune thrombocytopenia, autoimmune hemolytic anemia, and Guillain–Barre syndrome
[32][45]. Moreover, researchers have observed that AID represents a predisposed condition to COVID-19; indeed, researchers found a significantly higher prevalence of COVID-19 in AID patients and an increased COVID-19-related mortality in systemic sclerosis (SSc) patients’ subgroup
[33][46]. The researchers have also found that AID patients that are not on conventional synthetic disease-modifying anti-rheumatic drugs (mainly hydroxyl-chloroquine and methotrexate) report a higher prevalence of COVID-19; this suggests some protective role of these drugs against the most worrisome complications of COVID-19
[34][47]. AITD are organ-specific autoimmune diseases mediated by T helper (Th)1 lymphocytes, whose main clinical manifestations are GD and HT, which can respectively lead to thyrotoxicosis and hypothyroidism. Numerous environmental risk factors (such as drugs, stress, radiation, seasonality, smoking, viruses, and iodine) are considered triggers of AITD in individuals with a genetic predisposition. The viruses may play a crucial role in the AITD onset via the activation of the adaptive and innate immunity
[35][48]. Several viruses have been documented as a potential AITD culprit, such as the herpes simplex virus, Human T-cell lymphotropic virus-1, mumps virus, rubella, Epstein–Barr virus, enterovirus in HT, and retroviruses [human foamy virus or human immunodeficiency virus (HIV), human T-cell lymphotropic virus-1, and Simian virus 40] in GD
[36][49]. Moreover, the human parvovirus B19 (EVB19) and Hepatitis C virus (HCV) have been associated with the development of AITD
[35][36][37][38][48,49,50,51].
In fact, extrahepatic manifestations
[39][40][52,53], including Sjogren’s syndrome, mixed cryoglobulinemia (MC), and endocrinological diseases (AITD and type 2 diabetes), are diagnosed in 38–76% patients affected by chronic hepatitis C (CHC)
[41][42][54,55]. HCV seems to impair the self-tolerance (in immune cells or in thyrocytes) toward thyroid tissue, facilitating an autoimmune reaction against it in predisposed subjects
[43][44][45][46][47][48][49][50][56,57,58,59,60,61,62,63]. HCV thyroid infection increases the synthesis of CXCL10 in thyroid cells, with the subsequent attraction of more Th1 lymphocytes into the gland
[51][64]. Thyroid autoimmunity has also been reported significantly among MC + HCV patients versus the controls (AT 35 versus 16%; subclinical hypothyroidism, 11 versus 2%) (62). Notably, CHC patients, with or without MC but with AITD, showed an increased prevalence of PTC, suggesting that AITD might contribute towards TC development
[52][53][65,66].
There are conflicting results about thyroid dysfunctions or the new onset of thyroid autoantibodies among patients recovered from COVID-19. While several reports state that these phenomena are infrequent and should consider a routine reassessment thyroid function test (TFT) among patients with initially normal TFT as not necessary
[14][31][54][27,44,67], other researchers reported different results: in fact, Anaya and colleagues found that hospitalized patients with COVID-19 exhibited more frequent serologic thyroid autoimmunity than the pre-pandemic healthy controls (36.7% vs 20%
p 0.007), suggesting that SARS-CoV-2 may be a trigger for AITD
[55][68]. In another study, in 104 patients whose anti-thyroid antibody levels were remeasured after 3 months from their admission for COVID-19, increase in anti-thyroid peroxidase (AbTPO;
p < 0.001) and anti-thyroglobulin (AbTg;
p < 0.001) antibodies was observed, but not for anti-thyroid stimulating hormone receptor antibodies (
p = 0.486). Among 82 patients with negative anti-TPO findings at the baseline, a significant interval increase in anti-TPO titer (by >12 U) was observed in 16 subjects, of whom four became anti-TPO positive. In this sample subset, the high C-reactive protein during hospitalization (
p = 0.033), worse baseline clinical condition (
p = 0.018), and higher baseline anti-TPO titer (
p = 0.005) were associated with a significant increase in the anti-TPO titer. Nonetheless, it has been reported that 70% of this cohort of patients during the hospital stay have been treated with interferon beta-1b therapy, which likely confounded the investigation of the autoimmunity appearance along SARS-CoV-2-related thyroid dysfunctions
[56][69]. To date, several new cases of GD
[57][58][59][60][61][62][70,71,72,73,74,75], including with Graves’s ophthalmopathy (GO)
[57][70], and of HT
[62][63][64][75,76,77], following SARS-CoV-2 infection, have been reported in the literature. The time span between the infection and the onset of thyroid disease was heterogeneous, ranging from a concomitant appearance to more than 6 weeks, and all of these patients were managed with specific therapies. Several mechanisms have been hypothesized to explain the induction of autoimmunity elicited by SARS-CoV-2 infection: the immune system’s hyper-stimulation, the molecular mimicry between the self-antigens of the host and the virus, neutrophils extracellular traps, and finally, the virus induced the transcriptional changes in the immune genes (
Table 1). Conversely, AITD seem to be associated with the augmented risk of contracting the infection. In fact, to explore this hypothesis, researchers conducted, during the first phase of the pandemic (from April to September 2020), an observational study that involved 515 consecutive unselected patients affected by thyroid disorders, 25 of whom had, confirmed (11/25), or highly suspected (14/25) SARS-CoV-2 infection
[65][78]. The researchers separated the patients in two groups: patients with AITD (HT, Graves’ disease, patients with thyroid diseases, and circulating AbTPO and/or AbTg), and patients with no AITD. At the end of the survey, a higher prevalence of both symptomatic and asymptomatic COVID-19 was detected among the group of patients with AITD
[65][78].
Table 1.
Potential pathogenetic mechanisms of SARS-CoV-2 thyroid autoimmunity induction.