Avascular necrosis of the femoral head (ANFH) is a painful disorder characterized by the cessation of blood supply to the femoral head, leading to its death and subsequent joint collapse. Influenced by several risk factors, including corticosteroid use, excessive alcohol intake, hypercholesterolemia, smoking and some inflammatory disorders, along with cancer, its clinical consequences are thrombus formation due to underlying inflammation and endothelial dysfunction, which collaborates with coagulopathy and impaired angiogenesis. Nonetheless, angiogenesis resolves the obstructed free flow of the blood by providing alternative routes. Clinical manifestations of early stage of ANFH mimic cysts or lesions in subchondral bone, vasculitis and transient osteoporosis of the hip, rendering it difficult to diagnose, complex to understand and complicated to cure.
- avascular necrosis of femoral head
- impaired angiogenesis
- coagulopathy
- endothelial dysfunction
- bone disease
- skeletal abnormality
1. Introduction
2. Angiogenesis: Sprouting, Splitting and Stabilization
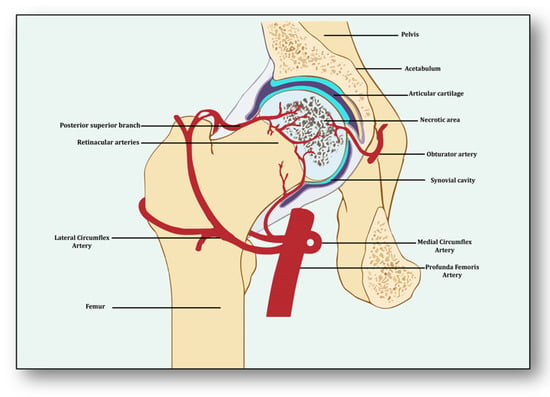
Angiogenesis is a process of developing new vessels from pre-existing vessels, and these vessels are considered to be the foremost organ in embryo development [5][15]. Vessels may develop, remodel and grow in different ways. They may sprout from already present vessels (sprouting angiogenesis) or develop by splitting from previously prevailing arteries or capillaries upon receiving the angiogenic stimuli (intussusceptive) [6][16]. They may enlarge and elongate from the coalescence of capillaries (coalescent angiogenesis), promoting the rapid expansion of the vasculature. Vessels may remodel themselves to increase their luminal diameter when more blood flow is required or there is a need to develop collateral bridges to provide alternative routes for the free flow of blood (arteriogenesis) [7][17]. Unlike angiogenesis, new blood vessels may be formed from the blood islands where no pre-existing vessels are present (vasculogenesis) [6][16]. All newly formed vessels must be mature and stable, otherwise they may be abnormal and leaking, further causing problems of local hematoma and edema [8][18]. These become stable due to the signaling molecules arranging pericytes overlaid on endothelial cells and the formation of the basement membrane, whereby junctions are established to allow optimum blood flow [9][19]. Angiogenesis has both beneficial and detrimental effects on health and disease, which makes it a potent hotspot for both pro-angiogenic and anti-angiogenic drug targets [10][20]. From the perspective of avascular necrosis, regulated angiogenesis is beneficial for collateral circulation to the necrotic area and its repair, whereas dysregulated angiogenesis is harmful [11][21]. The femur is a specially structured and highly vascularized bone, the longest in the human body. Its mechanical strength, recuperation, repair, regeneration and remodeling depend upon vascular health, which helps in supplying incessant blood and providing adequate oxygen, nutrients, growth factors and osteoprogenitor cells to the bone [12][22]. Consequently, angiogenesis is expected to revascularize, reperfuse and resorb the necrotic area. Branching from the circulatory system, the nutrient artery is the largest blood vessel that enters the medullary cavity and supplies almost half of the total blood volume to the femur [13][23]. At the proximal end, it forms anastomoses with perforating arteries, whereas at the distal side it merges with the profunda femoris artery. It extends longitudinally to the bone and divides into the lateral femoral circumflex artery and medial femoral circumflex artery [14][24]. Both branches of lateral femoral circumflex arteries feed the femur head region via lateral epiphyseal arteries and the neck region through posterior superior retinacular arteries [15][25]. The ligament of the femur head is also supplied by the anterior branch of the obturator artery of the hip bone, which traverses through the inferior part of the pubic ramus and anastomoses with the femoral artery and medial femoral circumflex artery [16][26] (Figure 1). With the advent of three-dimensional high-resolution imaging, a new aspect of the anatomy of vessels linking bone vasculature and bone marrow has been discovered [17][27]. This newly discovered vascular system comprises arterioles, venioles and capillaries, which collectively have been named as transcortical vessels (TCVs), which have shed light on the connection between endosteal and periosteal circulation [18][28]. A new subtype of blood vessels expressed from endomucin (Emcn) and a cluster of differentiation 31 (CD31) on endothelial cells in the bone known as type H has been identified lately [19][29]. Present in endosteum and metaphysis, this vessel is considered to be full of mesenchymal and osteoprogenitor cells, which mediate subchondral remodeling by coupling angiogenesis and osteogenesis [20][30]. Whether it contributes to ANFH pathology remains to be clarified as crosstalk between subchondral bone and articular cartilage during ischemia is unclear [21][31]. Obstruction or ischemia may take place in any of these arteries, but lateral and medial branches of the femoral circumflex or retinacular arteries are largely involved [22][32].3. Angiogenesis: A Predominant Pacifier in Avascular Necrosis
Several stimuli are received by endothelial cells from the local environment, prompting them to initiate angiogenesis [23][33]. These signaling stimuli augment endothelial cell activation and their migration along with apoptotic resistance, cytoskeletal reorganization and endothelial cell proliferation. Firstly, angiogenesis is triggered in response to ischemia, which is a dynamic process leveraging the equilibrium between pro-angiogenic and anti-angiogenic factors, resulting in the expansion of the vascular network [24][34]. Several vasculature and bone-derived angiogenic factors and stimulators play a role in angiogenesis to counterbalance the exigencies of nutrients and oxygen to the necrotic area [25][35]. They are hypoxia inducible factor-1α (HIF1-α), vascular endothelial growth factor (VEGF), vascular endothelial growth factor receptor (VEGFR), neuropilin 1 (NRP1), angiopoietin 1 (ANGPT1), angiopoietin 2 (ANGPT2), platelet-derived growth factor (PDGF), transforming growth factor-β (TGF-β), C-C motif chemokine ligand-2 (CCL-2), integrins αvβ3, αvβ5., vascular endothelial cadherin (VE-cadherin), cluster of differentiation 31 (CD31), plasminogen activators, inhibitor of DNA binding-1/inhibitor of DNA binding-3 (ID1/ID3), bone morphogenetic protein (BMP), prostaglandins (PTG), adenosine, pleiotrophin (PTN), delta-like canonical Notch ligand 4-Notch-Noggin (DLL4-NOTCH-NOG), receptor activator of nuclear factor-kappa β ligand/receptor activator of nuclear factor-kappa β/osteoprotegerin (RANKL/RANK/OPG), semaphorin (SEMA), nitric oxide (NO) and matrix metalloproteinases (MMPs) [26][36]. Nonetheless, molecular pathways between vasculature and bone interact and collaborate to initiate angiogenesis–osteogenesis coupling, which is required for the overall regeneration and repair of the necrotic area [27][37]. Endothelial cells along with pericytes initiate endocrine signaling, whereas osteoblasts and osteoclasts trigger angiogenesis to manage and maintain vasculature. After ischemia, hypoxic conditions emerge, which induce HIF-1α and VEGF in response [28][38]. It has been considered that HIF-1α is the precursor for the upregulation of VEGF, which is corroborated by the finding that transplantation of HIF1-α with transgenic bone marrow cells onto the necrotic area upregulated VEGF and increased angiogenesis, resulting in the repair of the necrotic area [25][35]. The nuclear signal transduction augments the translocation of HIF1-α to form a complex with HIF1-β and transcriptional co-activator E1A-associated protein/CREB binding protein (p300/CBP), which helps them to bind with the hypoxia response element [29][39]. It translates into the activation of several angiogenic genes such as VEGF, ANGPT-2 and nitric oxide synthase (NOS). VEGF plays the main role in bone remodeling via differentiation of osteoblasts and promoting endothelial cells at the affected area [30][40]. Besides the involvement of other forms of VEGF (VEGF-B, VEGF-C, VEGF-D and placenta growth factor), VEGF-A is primarily involved in angiogenesis and vasculogenesis during ischemic insult by binding and activating both VEGF receptors, i.e., VEGFR-1 and VEGFR-2, for vascular permeability, cell migration, vascular function and vessel maintenance [31][41]. Besides promoting endothelial cell differentiation, migration and proliferation, VEGF initiates the recruitment of bone-marrow-derived endothelial progenitor cells at the affected area [32][42] (Figure 2). Consequently, it promotes morphogenesis of the growth plate, blood vessel formation and remodeling of the affected cartilage [33][43]. The mechanism of VEGF-induced angiogenesis is essential in cartilage revascularization at both early stage and end stage after necrosis [34][44]. It is expressed in the edematous area of the necrotic zone and plays a significant role in the repair of the ongoing hypoxia-induced osteonecrotic area [35][45]. In the absence of VEGF, angiogenesis has been observed to be arrested, and the process of trabecular and cortical bone repair is significantly attenuated [36][46]. Moreover, it directly influences the osteoblast activity by increasing nodule formation and alkaline phosphatase, thereby promoting mineralization in a dose-dependent manner [37][47]. This suggests that the upregulation of VEGF in osteoblasts during hypoxia participates in and contributes to the healing process by promoting initial calcification at the site of injury [38][48].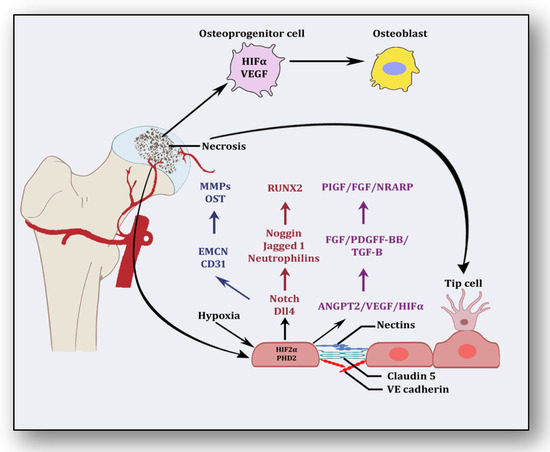
4. Coagulopathy: A Culprit Alliance of Thrombophilia and Hypofibrinolysis
Following the revelation from the first study by Hamilton et al. in 1965, several studies have endorsed that the pathology of osteonecrosis resulting from vascular ischemia is strongly influenced by coagulopathy [46][61]. Intravascular coagulation and thrombosis coupled with excessive thrombophilia and hypofibrinolysis are the major reasons [47][62]. Thrombophilia, sometimes called hypercoagulability, is an abnormality of the clotting mechanism which promotes thrombus formation within walls of blood circulatory vessels. Thrombophilia predominantly develops into deep venous thrombosis (DVT) and pulmonary embolism (PE), two chief reasons for cardiovascular morbidity and mortality. Both of these hypercoagulable conditions are termed venous thromboembolism (VTE). VTE deteriorates fibrinolytic machinery causing hypofibrinolysis, an abnormal condition whereby clot-resolving factors are dysregulated and clot-forming conditions are promoted. Fibrinolysis is the process of breaking down thrombus or clots and is strictly regulated by activators such as tissue plasminogen activator (tPA) and urokinase-type plasminogen activator (uPA) as well as inhibitors like tissue factor plasminogen inhibitor (TFPI) and plasminogen activator inhibitor-1 (PAI-1) and a fibrinolytic protease, plasmin. Plasminogen converts to plasmin via FXIa, FXIIa and kallikrein. This step triggers fibrinolysis by activating tPA within endothelial cells and uPA through the urinary epithelium, monocytes and macrophages. These factors play a significant role in breaking down and clearing clots from vasculature, whereas hypofibrinolysis (decreased levels of tPA and increased levels of PAI-1) impairs clot breakdown and prolongs its clearance. Several primary factors such as low levels of activated protein C (APC), protein S, factor V Leiden, activated protein C resistance (APCR), low levels of tPA or high levels of PAI-1, high levels of von Willebrand factor (vWF), high levels of lipoprotein(a) (Lp(a)) and homocystinuria along with secondary factors such as antiphospholipid antibodies, corticosteroid use, systemic lupus erythematosus (SLE) and caisson disease hemoglobinopathies, hemato-oncological diseases such as chronic myelogenous leukemia, acute lymphoblastic leukemia and multiple myeloma also participate in and contribute to causing hypofibrinolysis [47][48][49][50][51][52][53][54][55][62,63,64,65,66,67,68,69,70]. The coagulopathy cascade comprises a localized and speedy activation of inactive serine proteases (clotting factors) sequentially to generate thrombin resulting in clot formation (fibrin mesh) [56][71]. This pathway is triggered by sub-endothelial mural cells and fibroblasts of vascular adventitia. Coagulation may also trigger due to low levels of circulating polymorphonuclear neutrophils and monocytes/macrophages [57][72]. The first and foremost trigger is the exposure of tissue factor (TF) due to the severity of endothelial cell damage [58][73]. This exposed TF combines with factor VII to activate it to FVIIa, culminating in a sequence of activating factors such as FIX to FIXa and FX to FXa [59][7]. FXa turns prothrombin to thrombin, which further activates FV and FVIII to FVa and FVIIIa, a step responsible for converting prothrombin to thrombin by activating FX to FXa [59][7]. Furthermore, this thrombin-mediated fibrin clot is solidified by FXIa and interlinked by FXIIIa [60][74]. Activated platelets aggregate to form this clot as TF-presenting cells, ultimately augmenting coagulation and thrombus formation [61][75]. This process is simultaneously regulated by inhibitors of coagulation so that clot formation is not unnecessary and remains localized. TFPI, anti-thrombin (AT) and protein C are three major inhibitory molecules that check and resolve excessive coagulation within vessel walls [62][76] (Figure 3). Furthermore, the fibrinolytic pathway mediates vessel wall agility, integrity and healing.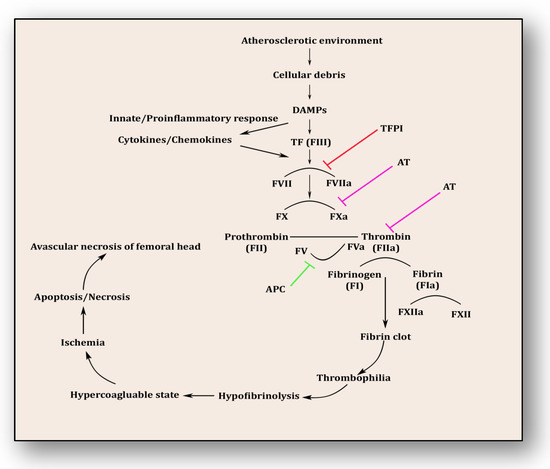
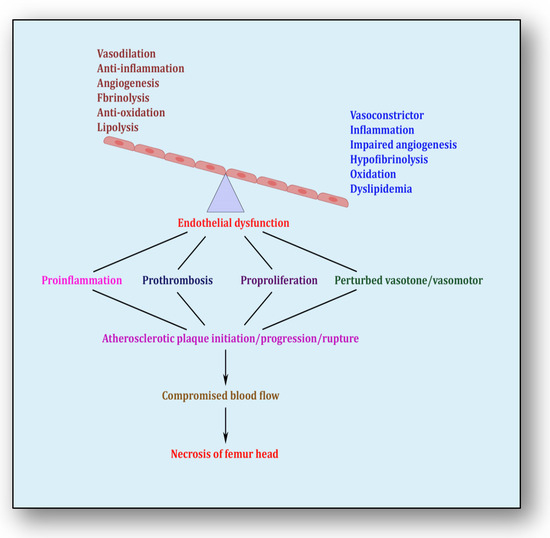
5. Endothelial Dysfunction: Holding Hands with Inflammation
The endothelium is a cell lining positioned on the inner surface of the blood vessels dividing circulating blood from the tissue. In response to various physical and chemical stimuli, such as perturbed blood flow, excessive intramural pressure, oxidative stress, cellular damage, high levels of homocysteine, hyperlipidemia, toxic chemicals and bacterial/viral infections, it initiates endocrine, paracrine and autocrine functions to produce vasodilators such as nitric oxide (NO), prostacyclin (PGI2) and endothelium-derived hyperpolarizing factors (EDHFs) and vasoconstrictors such as endothelin-1 (ET-1) and thromboxane-A2 (TXA2) [71][85]. The endothelium regulates homeostasis by maintaining the balance between vasodilators and vasoconstrictors, anticoagulants and procoagulants, inflammatory and anti-inflammatory molecules, oxidants and antioxidants as well as profibrinolytics and antifibrinolytics (Figure 4) [72][86]. Due to several risk factors, this homeostasis is lost and is termed endothelial dysfunction. In prolonged endothelial dysfunction, cholesterol microcrystals, monocytes and lymphocytes enter layers of endothelium and initiate inflammatory response, which helps in the formation of fatty streaks resulting in plaque setup, its progression and rupture. Plaque rupture expounds thrombus formation, which couples with coagulation cascade, resulting in atherogenesis and vascular ischemia [73][87]. Alluding to its contribution to the pathology of several diseases, endothelial dysfunction has been recognized as the diagnostic and prognostic marker for developing atherosclerotic plaque at all phases of initiation, progression and its worst outcomes of plaque rupture [74][88].