Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Yasuhiro Nishida and Version 3 by Camila Xu.
Astaxanthin (AX), a lipid-soluble pigment belonging to the xanthophyll carotenoids family, has recently garnered significant attention due to its unique physical properties, biochemical attributes, and physiological effects.
- astaxanthin
- microalgae
- mitochondria
- SDGs
- anti-aging
- slow-aging
1. Introduction
Astaxanthin (AX), a captivating red-orange pigment belonging to the carotenoid family, has garnered tremendous attention in recent years owing to its extraordinary physical properties, biochemical characteristics, and physiological effects. This remarkable compound has emerged as a promising contender in the realm of human health and well-being, prompting a surge in scientific research. In fact, the number of PubMed-indexed publications on astaxanthin has soared exponentially, skyrocketing from a mere 29 papers in 2001 to a staggering 414 papers in 2022, marking a fourteen-fold increase over the past decade. To date, more than 3500 papers on “astaxanthin” have been indexed in the PubMed database (Figure 1).
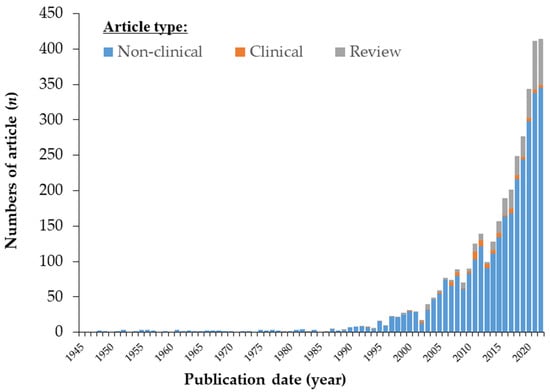
Figure 1. Number of scientific papers on astaxanthin (AX) by the end of 2022. Number of articles in PubMed (https://pubmed.ncbi.nlm.nih.gov/, accessed on 30 June 2023) by year. The keyword query “astaxanthin” was used to search the PubMed database. Note that “clinical trial” and “review” articles were selected as article type tags on PubMed, resulting in differences from the actual number of clinical reports shown in Section 3.1Section 3.3.1.1 of original paper.
The global landscape of AX research is undergoing a notable shift, with countries worldwide, including Japan, spearheading significant advancements. Initially relegated to a modest role as a coloring agent in the aquaculture and poultry industries, AX has experienced a remarkable transformation. As the fisheries, poultry, and livestock sectors underwent structural changes, the use of AX in commercial feeds expanded exponentially. Simultaneously, novel applications in human health have propelled AX to new heights of commercial significance. Intriguingly, recent studies have begun to unveil the potential of AX in addressing health challenges stemming from societal shifts.
According to available market research and forecasts, the market for AX and its end products is currently estimated to be valued between USD 647.1 million and USD 1633.7 million in 2021. Furthermore, revenue projections suggest that the market is expected to reach USD 965 million to USD 3200 million by 2026 [1][2][3][1,2,3]. This indicates a projected compound annual growth rate (CAGR) ranging from 8% to 16%. The rapid expansion of AX can be better comprehended by delving into its discovery, current applications, and how the latest research and market trends are shaping its future.
2. Nature and Cultural Aspects of Astaxanthin
2.1. Astaxanthin; Chemistry, History of Discovery and Structural Investigation
2.1.1. Astaxanthin; Chemical Structure and its Properties
Astaxanthin (3,3′-dihydroxy-β,β-carotene-4,4′-dione; AX) is a carotenoid with a chemical formula of C40H52O4 and a molecular structure that includes two hydroxyl and two carbonyl groups. AX exhibits an orange to deep-red color due to the presence of 13 conjugated double bonds. It is important to note that in its crystalline form, astaxanthin takes on a glossy black-purple color. The molecular structure of AX is symmetrical, with two chiral carbons at the 3 and 3′ positions of both terminal β-ionone groups, giving rise to three possible optical isomers (stereoisomers): (3S,3′S), meso (3R,3′S), and (3R,3′R)-AX. Additionally, due to the presence of nine double bonds in the polyene moiety, there can theoretically be 512 geometric isomers. While most naturally occurring AX is in the all-trans configuration, 9-, 13-, and 15-cis isomers have also been identified (Figure 2).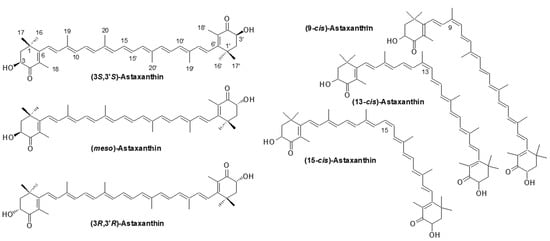
Figure 2.
Astaxanthin; structure, optical isomers and major geometric isomers.
2.1.2. Astaxanthin; Discovery and History of Structural Investigation
Investigations into astaxanthin (AX) began soon after the initial discovery of carotenoids. The study of carotenoids dates back to the early nineteenth century, when carotenoids were first found and extracted from paprika (in 1817), saffron (in 1818), annatto (in 1825), carrots (in 1831), and autumn leaves (in 1837) [5][6][5,6]. In those early years, carotenoid structures were still largely unknown, and their characterization was primarily based on their solubility and light absorption properties. AX seems to be the same as that initially called crustaceorubin by the British naturalist Henry Nottidge Moseley in 1877 and by Marian Isabel Newbigin in 1897 [7]. The early 20th century marked a major turning point in carotenoid analysis with the invention of chromatography, a revolutionary biochemical technique that became a staple in the chemistry of natural organic compounds. In 1906, Tswett successfully separated carotenes, xanthophylls, and chlorophylls from green leaves using column chromatography for the first time. Subsequently, the 1930s became known as the “golden age” of carotenoid structure elucidation. During this period, Karrer and Kuhn characterized eight carotenoids, including β-carotene, which they discovered to be a precursor of vitamin A. Their remarkable achievements earned them the Nobel Prize in Chemistry [8]. They also elucidated the structures of lutein, zeaxanthin, and AX [5][6][5,6]. At that time, carotenoid structural studies were conducted using elemental analysis and oxidative degradation reactions with strong oxidizers like KMnO4. However, these techniques were not sensitive and required several grams of carotenoids in crystalline form for analysis. In the 1970s, significant improvements in analytical instrumentation, including the introduction of various spectroscopic and separation techniques such as MS, 1H-NMR, 13C-NMR, and HPLC, revolutionized the analysis of carotenoids. These advancements made it possible to analyze smaller samples more effectively. As a result, over 600 carotenoids found in nature have been structurally elucidated [4][9][4,9]. The structure of AX was elucidated relatively early in the history of carotenoid structural determination. In 1933, Kuhn and Lederer isolated two carotenoids from the shell and eggs of the lobster species Astacus gammarus (now known as Homarus gammarus) and named them “astacin” (now known as astacene) and “ovoester” [10]. In 1937, Stern and Salomon isolated a protein complex called “ovoverdin” from lobster, and in 1938, Kuhn and Sörensen further characterized “ovoverdin” and identified “ovoester” as a xanthophyll carotenoid, renaming it “astaxanthin” [11][12][11,12]. The name “asta” is derived from “Astacus” the genus name of the lobster. Kuhn and Sörensen demonstrated that AX exhibited behavior consistent with ovoester based on its melting point (215.5–216 °C) and elemental analysis. Additionally, astacin was determined to be an oxidized artifact of AX. Based on these findings, the structure of AX was determined to be 3,3′-dihydroxy-β,β-carotene-3,3′-dione [11]. In 1933, von Euler et al. isolated the red pigment “salmon acid” from salmon muscle [13], and in 1973, Khare et al. showed that salmon acid was identical to AX based on MS and 1H-NMR spectral data [14]. The search for natural sources of AX during the 1948–1950s led to its extraction from flamingo wings [15], grasshoppers, and other insects [16], as well as from the flower petals of the Adonis plant [17]. These early works on the isolation and identification of AX were published in “Nature,” one of the most prominent scientific journals, which highlights the great interest in natural pigments at that time. Subsequently, as described in Section 2.2, AX was found to be widely distributed in microorganisms, algae, and animals. Since 1970, AX, like other carotenoids, has been characterized using various spectroscopic techniques, including MS, 1H-NMR, and 13C-NMR [4]. Furthermore, X-ray crystallography was conducted in the 2000s [18][19][18,19]. Figure 3 shows an Oak Ridge Thermal Ellipsoid Program (ORTEP) diagram of a single molecule obtained from a single crystal of all-trans AX.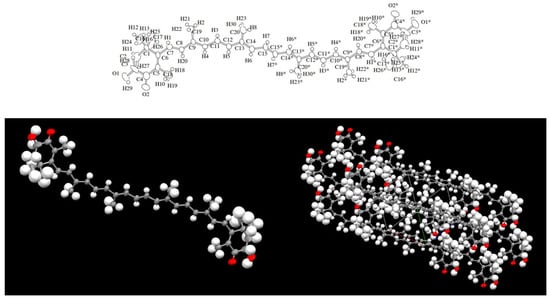
Figure 3. Oak Ridge Thermal Ellipsoid Program (ORTEP) diagram of a single molecule and the crystal structure obtained from a crystalline sample of all-trans astaxanthin. This figure was prepared based on reference [18].
2.1.3. The History of Astaxanthin Research in Japan
Today, AX research, including studies related to its biotechnology, is being conducted globally. However, in its early phases until the 1990s, it was predominantly carried out in Japan, where Japanese researchers made significant contributions to the field. Let us examine the history of AX research in Japan. In Japan, AX research initially began in the field of fisheries. In the 1970s, Matsuno et al. conducted extensive research on carotenoids found in various aquatic animals, approaching the subject from the perspectives of natural product chemistry and comparative biochemistry (for detailed information, refer to other reviews [20][21][22][20,21,22]). Furthermore, Hata, Katayama, et al. studied the pathway of AX production in goldfish (colored varieties of Carassius auratus), nishikigoi (colored varieties of Cyprinus carpio), and Japanese tiger prawn (Marsupenaeus japonicus, formerly Penaeus japonicus). They proposed the pathway of AX biosynthesis in these aquatic animals, considering the structures of various metabolic intermediates, using dietary carotenoids such as zeaxanthin and β-carotene [23][24][25][23,24,25]. Kitahara, Hata, Hatano, Ando, et al. also contributed research on the metabolism of AX in salmon [26][27][28][29][26,27,28,29]. In the 1980s, Matsuno, Fujita, and Miki et al. made further discoveries in AX research. They revealed the reductive metabolism of AX to tunaxanthin through their studies on the coloration of marine fish, such as sea bream and yellowtail, and the metabolism of carotenoids in their eggs [30][31][30,31]. Additionally, Miki et al. reported that the administration of AX to aquaculture fish improved egg quality, hatching, development, and growth of fry. These findings were based on their studies of AX dynamics in fish eggs [20]. In the 1990s, Miki et al. (1991) made a significant discovery regarding the antioxidant activity of AX. They found that AX exhibited a much stronger (more than 100 times) capability in quenching singlet oxygen than that of α-tocopherol (vitamin E) and that AX scavenged free radicals, superior to other examined carotenoids, including β-carotene and zeaxanthin, as well as α-tocopherol [32]. Furthermore, Nishino et al. conducted research on the anti-carcinogenic effects of AX and other carotenoids [33][34][33,34]. Prior to the 1990s, AX had primarily been studied in the field of fisheries. However, these findings sparked research on its applications in medicine and human health. Meanwhile, in 1993, the Marine Biotechnology Institute in Japan discovered an AX-producing marine bacterium, later identified as belonging to the genus Paracoccus [35]. Subsequently, in 1995, AX biosynthesis genes, including a novel key gene for the ketolation reaction [36], were isolated from this Paracoccus strain, and their functions were clarified by Misawa et al. [37], followed by the isolation of the key gene from the green alga Haematococcus pluvialis [38]. This breakthrough led to the development of current research not only in AXs biosynthesis but also in metabolic engineering and synthetic biology for AX production. The pioneering studies conducted in the 1990s laid the foundation for AX and carotenoid research as it stands today. Since 2000, AX has garnered significant attention in the field of preventative healthcare, particularly in relation to various lifestyle-related diseases. It has also gained recognition in the cosmetics industry for its anti-photooxidation and skin-aging effects. As a result, there have been a growing number of studies conducted worldwide on AX. In Japan, AX research and applications have been particularly prevalent in the field of ophthalmology. Considerable clinical evidence has accumulated regarding the effects of AX on eyestrain (asthenopia) [39][40][41][42][43][44][45][46][47][39,40,41,42,43,44,45,46,47]. Based on these research findings, several functional food products have been launched in Japan.2.1.4. Astaxanthin; Optical Isomers
In Section 2.1.1, it is mentioned that there are three possible optical isomers of AX. In the past, when AX was extracted from lobsters by Kuhn and Sörensen, it was considered optically inactive since it displayed minimal optical rotation [11]. In 1975, Liaaen-Jensen et al. successfully obtained optically active AX from the green alga Haematococcus pluvialis strain NIVA-CHL 9, now referred to as H. lacustris strain NIVA-CHL 9. In this study, H. lacustris will be referred to as Haematococcus algae, unless otherwise specified [48]. For further details, please see Section 2Section 2.2.2.2.2. Liaaen-Jensen et al. reduced the isolated AX from Haematococcus algae using NaBH4, and the resulting product exhibited a circular dichroism (CD) spectrum consistent with (3R,3′R)-zeaxanthin. Consequently, it was determined that AX derived from Haematococcus algae possesses a stereoconfiguration of (3S,3′S) [49][50][49,50]. Please note that the “R” and “S” nomenclature rules for absolute configuration were followed, where the hydroxyl group connecting the chiral carbon at the 3,3′ position to the chiral center is oriented upward (HO►), designating zeaxanthin as “R” and AX as “S”. In 1976, Andrewes et al. made a significant discovery regarding AX obtained from Phaffia yeast, specifically Phaffia rhodozyma (currently known as Xanthophyllomyces dendrorhous). They observed that the CD spectrum of AX from this yeast was completely opposite to that of (3S,3′S)-AX, indicating that the AX from Phaffia yeast exclusively adopts the (3R,3′R) conformation [51]. This finding prompted to conduct a meticulous comparison of the CD spectra of AX obtained from various marine animals. The observed differences in intensities (Δε) indicated that the AX from marine animals is a mixture of optical isomers. In 1979, Vecchi and Müller successfully separated racemic AX into three optical isomers using high-performance liquid chromatography (HPLC) in a normal-phase system. They employed diastereomeric esters of di-(−)-camphanate for this purpose [52]. Through this method, they were able to separate the optical isomers of AX obtained from lobster, shrimp, salmon, and starfish. The analysis revealed that AX in shrimp, salmon, and starfish comprised a mixture of the three optical isomers (3R,3′R), meso, and (3S,3′S). Furthermore, Vecchi and Müller directly separated the racemic AX into the three optical isomers using HPLC with a commercially available column called Sumichiral OA-2000. This column utilized an optically active stationary phase known as N-3,5-dinitrobenzoyl-D-phenylglycine (refer to Supplementary Figure S2B in original paperSupplementary Figure S2B) [53]. By employing this method, Vecchi and Müller confirmed the existence of three stereoisomeric forms of AX in various marine animals.2.1.5. Astaxanthin; Geometric Isomers
Most natural AX exists as a mixture of geometrical isomers, including small amounts of 9-cis, 13-cis, and 15-cis forms, along with the all-trans form (Figure 2). In 1980, Roche’s group isolated ten geometric isomers of AX by HPLC, and their UV-VIS and 1H-NMR spectral data were reported [54]. Very recently, Yao et al. reported studies on the Raman spectra of the isomers of AX using density functional theory (DFT) calculations [55]. They confirmed that the theoretically calculated Raman spectra accurately reproduced the experimentally recorded Raman spectra of the all-trans, 9-cis, and 13-cis isomers of AX. They expanded the theoretical studies to the other isomers of AX (15-cis, 9,9′-cis, 9,13-cis, 9,13′-cis, 9,15-cis, 13,13′-cis, and 13,15-cis isomers) and proposed the assignment of the vibrational modes. They also discussed the stability of the isomers by comparing the theoretically predicted relative energies and estimated that the ratio of the all-trans configuration is approximately 70%, while 9-cis and 13-cis isomers each account for about 10%. The other isomers make up less than 2% under thermal equilibrium conditions. Recently, Honda et al. introduced effective methods to generate geometrical isomers of AX in a thermally dependent process [56][57][56,57]. To date, there have been few reports on the physiological activities related to the geometric isomers of AX [58]. From a physicochemical perspective, several properties of cis isomers, including absorption maxima, solubility in solvents, and antioxidant activity, have been shown to differ from those of the all-trans form [4][54][58][59][60][4,54,58,59,60]. Specifically, the recent reports by Honda et al. and others indicate that certain geometric isomers may have higher bioavailability in rodents and are expected to have clinical applications in the near future [58][60][61][58,60,61].2.1.6. Astaxanthin Fatty Acid Esters
In addition to the free form, where no hydroxyl group modifications are present, AX also occurs naturally in a form where hydroxyl groups are modified by fatty acid esters (details of the distributions are described in Section 2.2). AX exists in both mono- and di-ester forms, with the ester moieties commonly composed of saturated fatty acids ranging from C12 to C18. Esterified derivatives of AX with highly unsaturated fatty acids such as eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) have also been reported in marine animals. For example, AX in Haematococcus algae occurs mainly as a series of monoesters with C16 to C18 fatty acids [62][63][64][62,63,64], while in krill it occurs as diesters with highly unsaturated fatty acids such as DHA and EPA [65][66][65,66]. Therefore, when quantification of AX is required, it is often calculated from the absorbance value based on the absorption coefficient of the free form as a tentative quantification value. For more accurate quantification, saponification should be applied, and the free AX content should be quantified by HPLC. In other words, the value obtained by converting all of the esterified AX into its free form is often used as the AX concentration. The reasons for this and the details of the analytical methods are discussed individually in Section 2Section 2.4.2.4.2. One of the most important concerns is that AX can be readily converted to “astacene” (3,3′-dihydroxy-2,3,2′,3′-tetradehydro-β,β-carotene-4,4′-dione) through oxidation in alkaline solutions in the presence of oxygen [67] [67]. Therefore, to accurately quantify AX, esterified AX must first undergo alkaline saponification under anoxic conditions or enzymatic treatment with cholesterol esterase from Pseudomonas sp. [67][68][67,68]. The enzymatic treatment is generally more convenient as the hydrolysis reaction proceeds without artifacts under normal oxygen levels.2.1.7. Astaxanthin Aggregates
Similar to many other carotenoids, AX is believed to undergo self-aggregation in hydrated polar solvents, resulting in the formation of aggregates [69][70][69,70]. The water concentration in the AX-solvent mixture influences the morphology of these aggregates and significantly impacts their photophysical properties. Spectroscopic analysis reports suggest that the absorption spectrum of AX aggregates is either blue-shifted (H-aggregate) or red-shifted (J-aggregate) compared to that of the monomer, reflecting the conditions during aggregation [71][72][73][71,72,73]. In the J-aggregate, astaxanthin molecules are arranged from head to tail, forming a relatively relaxed aggregate. Conversely, the H-aggregate exhibits a tighter “card-pack” stacking of polyene chains, which are somewhat aligned in parallel to each other [74]. Notably, the formation of H-aggregates is a unique characteristic of carotenoids possessing a hydroxyl group on the terminal cyclohexene ring. Introducing an O-R group in place of the hydroxyl group inhibits aggregation, thereby strongly suppressing aggregation in the ester form of AX [75]. Aggregates often exhibit significant differences in behavior compared to non-aggregated forms of biomolecules. These changes in physical properties can have a significant impact on their biological activity, particularly their pharmacological activity. For instance, H-aggregates of carotenoids demonstrate higher photostability in aqueous solutions compared to monomers; however, their radical scavenging activity and ability to quench singlet molecular oxygen are much lower than those of the monomers [75][76][75,76]. Incorporating biomacromolecules and amphiphilic compounds such as DNA, proteins (e.g., bovine serum albumin), and polysaccharides (e.g., arabinogalactan chitosan) can stabilize the formation of AX aggregates with biomacromolecules [75][77][78][75,77,78]. Recent studies have reported successful incorporation of H- and J-aggregates into DNA/chitosan co-aggregates and the preparation of complex nanosuspensions containing these two types of aggregates [77]. AX aggregates, which are typically unstable, can be stabilized by incorporating them into hydrophobic microdomains of these polymers, such as DNA/chitosan complexes, even in the absence of EtOH/water solvents. Highly aggregated molecular complexes have demonstrated different behavior in terms of radical scavenging activity compared to monomers in simple aqueous polar solvents [77]. This difference may be attributed to the newly formed intermolecular hydrogen bonds with the biopolymer and the presence of a π-π conjugated structure in the intermolecular association. These factors may explain the variation in antioxidant activity between H- and J-aggregates due to their different electron transport capacities [77]. However, there are still many unresolved aspects regarding the physiological activity of aggregates, and further investigations are required. Recently, researchers have isolated five distinct forms of AX aggregates that allow for the adjustment of intermolecular coupling between AX molecules. Time-resolved absorption spectroscopic studies with sub-30 fs time-resolution have been conducted on these aggregates [79]. Each form of AX aggregate is capable of undergoing intermolecular singlet fission, with rates of triplet generation and annihilation that can be linked to the strength of intermolecular coupling. This finding challenges the conventional model of singlet fission in linear molecules [80], as it demonstrates that the triplet state of AX is directly formed from the initial 1Bu+ (S2) photoexcited state through an ultrafast singlet fission process. This discovery highlights the potential use of AX aggregates, particularly the H-aggregate, as photoprotectors in biological systems. The H-aggregate of AX exhibits a significant hypsochromic shift in absorption, extending into the UV spectral region, compared to that of the monomer. Consequently, the H-aggregate of AX efficiently absorbs light in the UV—blue spectral range. Upon photoexcitation, the H-aggregate of AX can safely dissipate its energy as heat through the triplet excited state, which is formed via the ultrafast fission process. The significance of aggregates in natural systems is further discussed in Section 2.1Section 2.1.8.8.2.1.8. Carotenoproteins: Astaxanthin-Protein Complexes
AX has hydroxyl groups at the C3 and C3′ positions and carbonyl groups at the C4 and C4′ positions. Therefore, it exhibits a high affinity for certain proteins, such as albumin, and can readily form pigment-protein complexes. In many marine animals, AX is present in the form of protein-bound complexes. One of the most well-known examples of AX-protein complexes is seen in the blue, purple, and yellow hues of crustacean exoskeletons, which are predominantly derived from the AX-protein complex [81]. For more information on carotenoid-protein interactions in aquatic organisms, refer to other reviews [81]. The relationship between the structure of AX and color has been extensively studied, particularly in the case of “crustacyanins”, which contribute to the blue to purple coloration of lobster shells belonging to the species Homarus gammarus and H. americanus. Crustacyanins are members of the lipocalin superfamily of proteins, as deduced from the amino acid sequence of their subunits, which are hydrophobic ligand-binding proteins [82][83][84][85][86][82,83,84,85,86]. The multimeric α-crustacyanin (with a maximum absorption wavelength of approximately 630 nm) and dimeric β-crustacyanin (with a maximum absorption wavelength of approximately 580–590 nm), isolated from lobster shells, exhibit blue to purple colors. The structure of α-crustacyanin has been investigated through CD spectra and X-ray crystallographic analysis of its substructure, β-crustacyanin. α-crustacyanin is a large macromolecule with a molecular weight of approximately 320 kDa, consisting of eight pairs of heterodimeric β-crustacyanin units, which are themselves composed of heterodimers formed by two apocrustacyanins. Apocrustacyanins comprise five subunits: A1, C1, and C2, each with a molecular weight of approximately 21 kDa, and A2 and A3, each with a molecular weight of approximately 19 kDa. In lobsters (Homarus gammarus ), the major subunits of β-crustacyanin are A2 and C1 [83]. Consequently, there are 16 molecules of free AX within α-crustacyanin, as each apocrustacyanin associates stoichiometrically with an equal amount of AX. X-ray crystallographic analysis of β-crustacyanin has revealed three characteristics resulting from the binding of AX: elongation of the chromophore due to the 6-s-trans planar structure, hydrogen bonding between the C4, C4′ keto group and water, as well as histidine residues, and the close interactions of the two chromophores (AX). Since the usual maximum absorption wavelength (λmax) of the AX monomer is around 470 nm, both β- and α-crustacyanins exhibit a strong bathochromic shift in their absorption spectra as a result of the conformational change of the chromophore within the protein, resulting in a purple and blue color, respectively [84]. Further bathochromic shifts of up to 45 nm can be observed due to aggregation effects during the association of β-crustacyanin with α-crustacyanin in lobster shells. In crustaceans, the combination of apoprotein subunits varies among species and mutations, contributing to the variation in the coloration of crustacyanins [86][87][86,87]. The relationship between the structure of AX and color has been extensively studied, particularly in the case of “crustacyanins”, which contribute to the blue to purple coloration of lobster shells belonging to the species Homarus gammarus and H. americanus. Crustacyanins are members of the lipocalin superfamily of proteins, as deduced from the amino acid sequence of their subunits, which are hydrophobic ligand-binding proteins [82][83][84][85][86][82,83,84,85,86]. The multimeric α-crustacyanin (with a maximum absorption wavelength of approximately 630 nm) and dimeric β-crustacyanin (with a maximum absorption wavelength of approximately 580–590 nm), isolated from lobster shells, exhibit blue to purple colors. The structure of α-crustacyanin has been investigated through CD spectra and X-ray crystallographic analysis of its substructure, β-crustacyanin. α-crustacyanin is a large macromolecule with a molecular weight of approximately 320 kDa, consisting of eight pairs of heterodimeric β-crustacyanin units, which are themselves composed of heterodimers formed by two apocrustacyanins. Apocrustacyanins comprise five subunits: A1, C1, and C2, each with a molecular weight of approximately 21 kDa, and A2 and A3, each with a molecular weight of approximately 19 kDa. In lobsters (H. gammarus ), the major subunits of β-crustacyanin are A2 and C1 [83]. Consequently, there are 16 molecules of free AX within α-crustacyanin, as each apocrustacyanin associates stoichiometrically with an equal amount of AX. X-ray crystallographic analysis of β-crustacyanin has revealed three characteristics resulting from the binding of AX: elongation of the chromophore due to the 6-s-trans planar structure, hydrogen bonding between the C4, C4′ keto group and water, as well as histidine residues, and the close interactions of the two chromophores (AX). Since the usual maximum absorption wavelength (λmax) of the AX monomer is around 470 nm, both β- and α-crustacyanins exhibit a strong bathochromic shift in their absorption spectra as a result of the conformational change of the chromophore within the protein, resulting in a purple and blue color, respectively [84]. Further bathochromic shifts of up to 45 nm can be observed due to aggregation effects during the association of β-crustacyanin with α-crustacyanin in lobster shells. In crustaceans, the combination of apoprotein subunits varies among species and mutations, contributing to the variation in the coloration of crustacyanins [86][87][86,87]. Another carotenoprotein that forms the exoskeleton in lobsters, similar to crustacyanin, is crustochrin. Crustochrin exhibits a yellow hue with hypsochromically shifted bands, having a maximum absorption wavelength of 400–410 nm. This protein contains approximately 20 astaxanthin molecules and demonstrates typical exciton-exciton interactions through natural H-aggregates (see Section 2.1.7), with the chromophores arranged in a stack-of-cards formation [88][89][88,89]. Interestingly, these two distinct groups of proteins (crustacyanins and cristochrins), in terms of color hue, have been found to localize differently within the lobster exoskeleton [86][90][86,90]. This characteristic localization will be described in Section 2.2.4. Recent studies indicate that crustacyanins are restricted to Malacostraca crustaceans but are widely distributed within this group. These crustacean-specific genes are divided into two distinct clades within the lipocalin protein superfamily. The fact that the crustacyanin gene family emerged early in the evolution of Malacostraca crustaceans suggests that this protein played a significant role in the evolutionary success of this group of arthropods [86][91][86,91]. Crustaceans, in particular, are known for their diverse species-specific shell colors and patterns, and these proteins are believed to be involved in functions such as protection through cryptic coloration, reproduction, and communication [92][93][92,93]. Moreover, in crustaceans, AX-binding proteins are not limited to α- or β-crustacyanin alone but also exist as complexes bound to “ovoverdins” and other proteins in crustaceans. Ovoverdins, reported as the pigment responsible for the dark green color (λmax; ca. 465–470 and 660–670 nm) of lobster ovaries and eggs [12][94][95][96][12,94,95,96], are a complex of AX (mostly in the free form) and lipovitellin, which is a predominant glycolipoprotein found in the yolk of egg-laying organisms [97]. Corresponding to their λmax, AX may bind to two distinct sites: one might be a weak non-specific association, and the other is a specific stoichiometric association with lipoproteins. Additionally, there are at least two different molecular-weight proteins (ca. 700 kDa and 600 kDa) [96]. In the latter, ovoverdin seems to form a multimer of four subunits consisting of a, b, c, and d, according to the SDS-PAGE results [96]. Similar AX-protein complexes have also been reported to form in other organisms. For example, a blue AX-protein complex called “velellacyanin” has been isolated from the blue mantle of the blue-colored “by-the-wind-sailor” jellyfish Velella velella [82][98][82,98]. There are two types of this protein, named V600 (λmax; ca. 600 nm) and V620 (λmax; ca. 620 nm), respectively, based on their maximum absorption wavelengths. The molecular weight of each is >300 kDa [82][98][82,98]. The velellacyanins are multimeric formations of multiple subunits and form a helical structure. The quaternary structures of velellacyanin and the N-terminal peptide sequence of the subunit comprising V600 reveal similarity to apocrustacyanin C [99][100][101][99,100,101]. In fact, immunocross-reactivity showed reactivity with polyclonal antibodies for not only apocrustacyanin C but also apocrustacyanin A [102]. However, it currently remains unclear whether these velellacyanin apoproteins belong to the lipocalin superfamily, and future studies, including their origin and evolutionary position, are expected. In echinoderms, two well-known carotenoproteins, as shown below, have been partially characterized by X-ray structural and CD spectral analyses; however, no phylogenetic or functional analysis of the proteins, including detailed genetic background, has yet been available. The common starfish Asterias rubens has “asteriarubin”, a purple-blue carotenoprotein, also present [103][104][103,104]. Asteriarubin (λmax; ca. 570 nm) is approximately 43 kDa and comprises four subunits with a molecular weight of approximately 11 kDa each. The major carotenoids in this protein are AX and its acetylenic and dehydro analogues, such as 7,8-dideoxyhydroastaxanthin and 7,8,7′,8′-tetradehydroastaxanthin. These carotenoids are metabolites of AX found in echinoderms and are described in Section 2.2Section 2.2.4.4. Interestingly, the amino acid sequence shows no homology to the apocrustacyanin subunits [81][103][81,103]. Reconstitution studies revealed similarities between the binding requirements of asteriarubin and crustacyanin; however, the tetrameric asteriarubin contains only one carotenoid molecule, and the CD spectrum shows no exciton splitting. Therefore, in this case, molecular aggregation and carotenoid-protein chromophore interactions were considered unlikely to be determinants of the bathochromic shift [81]. This slight bathochromic shift may be attributed to the absence of exciton effects compatible with extended π conjugation due to hydrogen bonding between the terminal polar group and the protein and the co-planarity of the ring [104]. Another type of carotenoprotein in echinoderms, the vivid blue skin of calcified starfish called “blue star”, Linckia laevigata, has a blue carotenoprotein called “linckiacyanin” (λmax; 395, 612 nm), with (3S,3′S)-AX as the major carotenoid [105]. Although the molecular weight of linckiacyanin is quite large (>103 kDa), the main glycoprotein subunit is small, at only approximately 6 kDa. However, the minimum molecular weight of the native subunits (approximately 16 kDa) means that there are at least 200 carotenoid molecules per molecule of linkyacyanin [105]. Since linkyacyanin showed no cross-reactivity with polyclonal antibodies for the β-crustacyanin subunit [102], it is possible that linkyacyanin is a distinct family member from the lipocalin superfamily. In Asia, it is easy to find vivid pink egg clumps on the surface of rice plants and on the walls of aqueducts for rice fields. This pigment is known as “ovorubin”. Ovorubin is a carotenoprotein found in the perivitelline fluid that surrounds the embryo of the fertilized egg, which is an accessory gland of the female reproductive tract of the South American freshwater snail Pomacea canaliculata (Gastropoda: Ampullariidae). Ovorubin is described as a large red AX-binding glycoprotein of approximately 330 kDa [106], and the binding of (3S,3′S)-AX and their fatty acid esters to ovorubin results in a small bathochromic shift (20–30 nm) to λmax 510 nm [81]. The protein is also an oligomer composed of three subunits of approximately 28, 32, and 35 kDa and is a very high-density glycosylated lipo-carotenoprotein (VHDL) with phospholipids, sterols, and carotenoids as ligands. It is highly glycosylated. This protein provides the egg with resistance against sun radiation and oxidation of lipids [107] and is thought to play an important physiological role in the storage, transport, and protection of carotenoids during snail embryogenesis [108]. In addition to ovorubin, there is another carotenoprotein called “alloporin” (λmax; 545 nm) found in the soft coral Allopora californica. Alloporin is approximately 68 kDa and comprises four subunits with a molecular weight of approximately 17 kDa each. It has an equal molar of (3S,3′S)-AX bound to it [109][110][109,110]. This seems to be similar to asteriarubin. The authors eagerly anticipate a future where the mysteries behind the physical-chemical properties and mechanisms of the chromophores found within these remarkable AX-containing carotenoproteins are unraveled. Aiming to shed light on the intricate details and functions of these chromophores, their characterization holds the key to unlocking a deeper understanding of the captivating structures and extraordinary roles played by these carotenoproteins. The journey to uncover their secrets promises to be a captivating exploration into the realms of science and discovery. All photosynthetic plants utilize carotenoid-binding proteins as an important component of their photosynthetic function. For example, in chloroplast thylakoid membranes, pigment molecules such as carotenoids and chlorophyll function within the light-harvesting protein complex (LHC), an antenna pigment-protein complex bound to the photosystem, to achieve extremely high-efficiency light harvesting. The photosystem II supercomplex (PSII) is also a pigment-protein complex that catalyzes water splitting and oxygen-evolving reactions in photosynthesis, converting light energy into chemical energy. The PSII core complex is composed of more than 20 subunits and contains approximately 35 chlorophylls (Chl) and 12 β-carotene molecules, as well as other oxidation-reduction cofactors required for electron transfer [111][112][111,112]. PSII is especially sensitive to light-induced damage (photodamage) among the components of photosynthesis because it is an extremely oxidative reagent with a high enough potential for the light-excited P680 Chl molecule, which is utilized in the MnCaO5 cluster to split water. If the rate of photodamage exceeds the rate of repair under excessively intense light conditions, a phenomenon called photoinhibition of PSII occurs. Photoinhibition of PSII is caused by reactive oxygen species (ROS) that are generated during the excitation energy transfer and electron transport processes. In particular, the repair system of photodamaged PSII is sensitive to various ROS. To reduce the effects of photoinhibition of PSII, plants have developed systems to quench or suppress the production of ROS. Non-photochemical quenching (NPQ) is one of these defense mechanisms in plants. One of the most important mechanisms is the involvement of carotenoids, which directly quench singlet oxygen generated by excited chlorophyll, or xanthophylls, which enhance heat dissipation of excess light energy through the xanthophyll cycle. Thus, it is clear that carotenoids play a pivotal role in oxygen-evolving photosynthesis. These carotenoids are present in membrane proteins in the thylakoid membrane or directly in the thylakoid membrane [111][112][111,112], while some coexist with water-soluble proteins. One of those groups is the orange carotenoid proteins, which are widely distributed in cyanobacteria [113]. However, there is no direct evidence that AX in the specific protein complex is involved in naïve plants’ photosynthetic function; however, certain green plants have AX-binding proteins as a photoprotector. For instance, the microalgae Coelastrella astaxanthina Ki-4 (Scenedesmaceae sp. Ki-4), isolated from the asphalt surface in mid-summer, benefits from a water-soluble AX-binding carotenoprotein called “AstaP” [114]. AstaP (AstaP-orange1) is a secreted protein that exhibits thermally stable 1O2 quenching activity [114], which is induced through exposure to strong light. The deduced N-terminal amino acid sequence of AstaP reveals that it represents a new class of carotenoid-binding proteins homologous to the fasciclin family proteins, with extensively N-glycosylated regions [114]. A related green alga, Scenedesmus sp. Oki-4N, has three comparable AstaP orthologs. However, AstaP-pinks has no glycosyl residues, while AstaP-orange2 has a glycosylphosphatidylinositol (GPI) anchor motif and a higher isoelectric point (pI = 3.6–4.7), which is significantly different from the original AstaP-orange1 (pI = 10.5) [115]. Orthologues of AstaP have also been found in diverse green algae, including Chlamydomonas reinhardtii and Chlorella variabilis, which are also induced by light irradiation [116]. These results are unique examples of how the use of water-soluble AX in photosynthetic organisms is a novel strategy to protect cells from severe photooxidative stress. The native form of AstaP (AstaP-orange1) from C. astaxanthina Ki-4 binds to AX quite specifically, whereas the expression of a correctly folded recombinant protein in E. coli and the evaluation of its binding to the protein revealed that it is also capable of binding to other xanthophylls and carotenes [117]. Recombinant AstaP is a ~20 kb water-soluble protein that accepts carotenoids in acetone solution or embedded in biological membranes. It then has the property of forming carotenoid-protein complexes with apparently equal stoichiometry [117]. Recently, recombinant plants have been developed that possess ketocarotenoids. These ketocarotenoids are typically lacking or completely absent from the photosynthetic system found in higher plants. However, it appears that they have been successfully integrated into the photosynthetic system. The high accumulation of AX and other ketocarotenoids has been found to impact growth, CO2 assimilation, and photosynthetic electron transfer in transgenic plants. Moreover, studies have demonstrated that ketocarotenoids act specifically on the thylakoid membrane and, more specifically, on PSII [118][119][118,119]. Interestingly, transplastomic lettuce, which has been genetically modified in its chloroplast genome, has been found to predominantly accumulate AX [120]. Despite having low levels of naturally occurring photosynthetic carotenoids [111], this lettuce exhibits normal growth similar to that of non-modified plants. Initially, the quantum yield of PSII in this lettuce is low under normal growth conditions; however, it becomes comparable to control leaves under higher light intensities. In AX-accumulating lettuce, in addition to β-carotene, echinenone and canthaxanthin are bound to the PSII monomer, while the normal binding of photosynthetic carotenoids is absent. This lack of normal carotenoid binding affects the assembly, photophysical properties, and function of PSII. However, the repair mechanisms in AX-accumulating lettuce enable the maintenance of PSII function despite photodamage. The high antioxidant capacity of AX, its esters, and other ketocarotenoids accumulated in thylakoid membranes is believed to provide protection against reactive oxygen species (ROS) generated during oxygenic photosynthesis [111]. When carotenoids and xanthophylls are highly accumulated, they can influence the physical properties of the lipid membrane itself, such as fluidity and permeability to small molecules [121]. In AX-accumulating lettuce, the high accumulation of xanthophylls may prevent the permeation of singlet oxygen produced by PSII and enable efficient quenching within the lipid bilayer. Specifically, AX and carbonyl derivatives of lutein have been observed to adhere to the surface of the PSII core complex, indicating their effective quenching of singlet oxygen (1O2). Essentially, the apparent photosynthetic capacity of this lettuce may be attributed to the antioxidant effect of AX and its derivatives, compensating for the absence of essential naturally occurring carotenoids [111]. The Silkworm, Bombyx mori larvae, possesses a carotenoid-binding protein (BmCBP) in their silk glands. This protein has an apparent molecular weight of 33 kDa and binds carotenoids in a 1:1 molar ratio. Lutein constitutes ninety percent of the bound carotenoids, although it also binds to other carotenoids [122]. In its cytoplasmic form, BmCBP acts as a non-internalized lipophorin receptor, binding to lutein and functioning as a “transport carrier” [123]. The deduced amino acid sequence of BmCBP indicates its affiliation with the steroidogenesis acute regulatory protein (StAR) family, featuring a distinctive structural element known as the StAR-related lipid transfer domain. This domain facilitates lipid translocation and recognition [122]. BmCBP demonstrates the ability to bind various carotenoids, including dietary AX, and transport them to the silkworm cocoon [124]. In recent years, significant advancements have been made, including the successful heterologous expression of BmCBP in E. coli while maintaining its functions [125]. Furthermore, the crystal structure of BmCBP has been determined through X-ray structural analysis, and the binding site for xanthophylls has been identified [126]. In recent years, carotenoid-binding proteins, including AstaP from the carotenoid binding protein (CBP) group, have emerged as promising antioxidant nanocarriers with potential applications in biomedicine [127]. The binding ability of AstaP to carotenoids also indicates its potential industrial utility in the recovery and concentration of carotenoids from crude extracts [125]. AX-protein complexes have also been observed in fish. In salmon, for instance, muscle AX exhibits specific binding to the surface of actomyosin, a protein present in skeletal muscles. Interestingly, the binding between this protein and AX does not appear to be stereoselective, meaning it does not show preference based on the spatial arrangement of AX molecules [128][129][128,129]. In general, animals are unable to synthesize carotenoids de novo, with a few exceptions, as mentioned in Section 2Section 2.2.4.2.4. Therefore, recent research has highlighted the involvement of transport proteins in the absorption of carotenoids from the intestinal tract and their transfer to various tissues. These transport proteins likely share functions with other carotenoids and lipids, such as cholesterol. However, the precise binding modes between these transport proteins and AX remain unclear, although it is presumed that the binding is relatively loose. It is still uncertain whether the binding occurs in a stoichiometric manner or not. Additionally, enzymatic degradation of carotenoids takes place in various tissues. This degradation involves reactions catalyzed by carotenoid cleavage oxygenases, resulting in the formation of apocarotenoids. A well-known example is the conversion of β-carotene into vitamin A retinoids by the enzyme BCO1. The detailed roles of these processes are discussed in other sections, specifically in Section 2.3.4 and SectionSection 4.1 4.1 of original paper for or mammals.2.1.9. Astaxanthin as a Powerful Antioxidant
AX is believed to exert its antioxidant activity through both direct quenching and scavenging of reactive chemical species, such as reactive oxygen species (ROS) and reactive nitrogen species (RNS). Additionally, it employs indirect mechanisms by inducing a group of antioxidant enzymes in biological systems. However, this research specifically focuses on the ROS-scavenging mechanism of AX.Quenching Singlet Oxygen
Singlet molecular oxygen (1O2) is generated from the ground-state triplet molecular oxygen (3O2: 3∑g−) through photochemical processes in biological systems [130][131][132][133][134][135][136][137][138][139][140][131,132,133,134,135,136,137,138,139,140,141]. Approximately 1O2 exists in two singlet states with different spins (1∑g+ and 1∆g), with 1∆g being the lowest excited singlet state. The former has a very short lifetime, while the latter, although short-lived, has a longer lifetime than the former. Therefore, the term “singlet oxygen” generally refers to the 1∆g state. The lifetime of singlet oxygen is also strongly influenced by the surrounding environment. Additionally, there is a very short-lived dimol molecule (O2(1∆g)-O2(1∆g)) that forms as a result of the reaction between two singlet oxygen molecules. This dimol molecule can be detected through luminescence in the red spectral region, which corresponds to twice the energy of O2(1∆g) emission in the infrared spectral region around 1270 nm. Under normal environmental conditions, the electric dipole transition of oxygen molecules from the ground triplet state (3Σg−) to the lowest electronically excited singlet state (1Δg) has an extremely low transition probability. This transition is forbidden due to considerations of spin angular momentum, orbital angular momentum, and parity. As a result, singlet oxygen is typically generated through interaction with photosensitizers such as porphyrins and chlorophylls. Additionally, it is believed that singlet oxygen can be generated in the absence of light through the Haber-Weiss reaction involving superoxide (O2•–) and hydrogen peroxide (H2O2). Furthermore, an autocatalytic reaction involving the cyclization of peroxyl radicals can also produce singlet oxygen via a tetraoxide intermediate. Therefore, the Russell mechanisms facilitate the generation of singlet oxygen from lipid peroxyl radicals. Enzymatic reactions, such as those catalyzed by myeloperoxidase in monocytes, can also lead to the production of singlet oxygen [130][137][138][140][131,138,139,141]. The production of singlet oxygen is indeed harmful to biological tissues. This is due to its ability to readily oxidize and modify lipids, proteins, and nucleic acids, which are vital for biological functions, thereby causing their loss of function through Diels-Alder reactions. Consequently, singlet oxygen has been implicated in several diseases. For instance, it has been strongly suggested that singlet oxygen is involved in light-exposed skin and ocular tissues, contributing to conditions such as skin aging, skin cancer, Porphyria, Smith-Lemli-Opitz syndrome, glaucoma, cataracts, and age-related macular degeneration. Moreover, even in the absence of light exposure, singlet oxygen’s involvement is strongly suspected in the onset or exacerbation of diabetes mellitus and bronchial asthma [141][142]. Carotenoids, such as lycopene, β-carotene, lutein, and AX, are highly efficient quenchers of singlet molecular oxygen (1O2). Their reaction rate constants, typically around 1010 M−1 s−1, approach the limit of diffusion control in solvents [76][142][76,143]. The quenching mechanism of 1O2 by carotenoids has been recently analyzed through quantum dynamics calculations and ab initio calculations [143][144]. Theoretical studies suggest that the ground-state singlet carotenoid (1Car) and 1O2 molecules can form a weakly bound complex, facilitated by the donation of electron density from the carotenoid’s highest occupied molecular orbital (HOMO) to the πg* orbitals of 1O2. The quenching of 1O2 is governed by a Dexter-type superexchange mechanism involving charge transfer states (Car•+/O2•−). Quantum dynamics calculations demonstrate that the quenching of 1O2 by carotenoid/O2 complexes occurs rapidly, within sub-picosecond timescales, due to strong electronic coupling. This theoretical study highlights the crucial role of carotenoid cation radical species (Car•+) in achieving efficient 1O2 quenching. Notably, AX is known for its high activity and stability against 1O2 quenching [142][143]. Experimental evidence supports the theoretical understanding that as the polyene chain length of carotenoids increases, i.e., the conjugated π-electron system becomes more extended, the HOMO level of carotenoids decreases, and their 1O2 quenching activity becomes stronger. For instance, Conn and Edge et al. conducted an evaluation of the singlet oxygen scavenging activity of various β-carotene and lycopene analogs with different lengths of conjugated π-electron systems. They extended the conjugated double bonds from 7,7′-dihydro-β-carotene (n = 7) to dodecapreno-β-carotene (n = 19), and the experimental results demonstrated that the 1O2 quenching activity increased as the length of the conjugated double bond was increased (refer to Table 1) [144][145][145,146]. AX (n = 13) possesses two expanded π-electron systems due to the presence of C4, C4′ diketo groups connected to the n = 11 conjugated polyene of C40 carotenoids. Additionally, AX has polar hydroxyl groups at both ends (refer to Figure 2). These molecular frameworks might specifically contribute to the expression of AXs superior antioxidant activity while maintaining its molecular stability. Comparisons of the activity of cis-isomers, such as β-carotene, with the all-trans geometrical isomer have been reported. Specifically, in terms of 1O2 scavenging activity, the all-trans configuration is known to exhibit the highest activity, followed by 15-cis and 9-cis isomers. This indicates that the rate constant for deactivating 1O2 by the cis-isomers decreases as the cis-bond of β-carotene moves away from the center of the molecule, resulting in less efficient 1O2 quenching compared to the all-trans isomers [144][145]. Time-resolved resonance Raman studies have shown that all β-carotene isomers share a common triplet state, characterized by a twist in the central carbon-carbon double bond compared to the ground state. This structural variation may have an impact on the conjugated system [146][147][147,148]. In biological model membranes, such as phospholipid liposomes, several studies have investigated the effectiveness of AX in inhibiting 1O2-induced peroxidation of phospholipid membranes. However, the observed activity of AX in these studies sometimes falls below expectations, such as exhibiting a lower reaction rate constant compared to β-carotene, which can vary depending on the detection system [148][149]. Nevertheless, when evaluated based on endoproducts as outcomes, AX has been found to exert effective protective actions against phospholipid membrane peroxidation and cellular damage induced by photosensitization in the presence of photosensitizers [149][150][151][152][153][150,151,152,153,154]. The reaction between AX and singlet oxygen is primarily attributed to physical quenching, as described previously. However, a small fraction of AX does form reactive products with 1O2. According to Nishino et al., the major products generated from this reaction are endoperoxides, specifically astaxanthin 5,6-endoperoxide or astaxanthin 5,8-endoperoxide. These endoperoxides represent 1O2 adducts formed at the C=C bonds of the β-end group [154][155].Table 1.
Singlet oxygen quenching activity of astaxanthin: comparison with common antioxidants [k(10
9
M
−1
s
−1
)].
| 1O2 Generator | EDN * | EDN * | NDPO2 * | EP * | |||
|---|---|---|---|---|---|---|---|
| Reference | [155][156] | [156][157] | [157][158] | [158][159] | |||
| Detection | Luminescence | Luminescence | Luminescence | Absorbance of DPBF | |||
| Solvent | CDCl3 | CDCl3/ CD3OD (2:1) |
DMF/ CDCl3 (9:1) |
CDCl3 | CDCl3/ CD3OD (2:1) |
EtOH/CHCl3 /D2O (50:50:1) |
EtOH/CHCl3/D2O (50:50:1) |
| 1. Carotenoids | |||||||
| Astaxanthin | 2.2 | 1.8 | 5.4 | 2.2 | 1.8 | 24.0 | 11.7 |
| Canthaxanthin | 2.2 | 1.3 | 2.0 | - | 1.2 | 21.0 | |
| Zeaxanthin | 2.0 | 0.73 | 3.4 | 1.9 | 0.12 | 10.0 | 11.2 |
| β-Cryptoxanthin | 2.0 | 0.27 | 1.7 | - | - | 6.0 | 7.0 |
| β-Carotene | 2.2 | 0.28 | 1.1 | 2.2 | 0.049 | 14.0 | 10.8 |
| Lycopene | 3.0 | 1.4 | 3.4 | - | - | 31.0 | 14.0 |
| Capsanthin | - | - | - | - | - | - | 12.1 |
| Lutein | 0.61 | 0.26 | 2.1 | 0.8 | - | 8.0 | 8.1 |
| α-Carotene | 0.66 | 0.23 | 0.93 | - | - | 19.0 | 10.0 |
| Fucoxanthin | 0.29 | 0.075 | 0.97 | - | 0.005 | - | - |
| Tunaxanthin | - | - | - | 0.15 | - | - | - |
| 2. Vitamin C | |||||||
| L-Ascorbic acid | - | - | 0.00089 | - | - | - | - |
| 3. Vitamin E | |||||||
| α-Tocopherol | 0.02 | 0.0039 | 0.049 | - | - | 0.28 | 0.13 |
| β-Tocopherol | - | - | - | - | - | 0.27 | 0.093 |
| γ-Tocopherol | - | - | - | - | - | 0.23 | 0.084 |
| δ-Tocopherol | - | - | - | - | - | 0.16 | 0.041 |
| Trolox | - | - | 0.010 | - | - | - | 0.042 |
| 4. Polyphenols/other phenolic antioxidants | |||||||
| α-Lipoic acid | 0.056 | 0.038 | 0.072 | - | - | 0.13 | 0.0019 |
| Ubiquinone-10 | 0.0019 | 0.0021 | 0.0068 | - | - | - | 0.062 |
| BHT | - | - | 0.004 | - | - | - | - |
| Caffeic acid | - | - | 0.0023 | - | - | - | 0.00069 |
| Ferulic acid | - | - | - | - | - | - | 0.00027 |
| CurcuminI | - | - | 0.0036 | - | - | - | - |
| (-)-EGCG | - | - | 0.0096 | - | - | - | 0.0051 |
| Gallic acid | - | - | 0.0023 | - | - | - | - |
| Pyrocatechol | - | - | 0.0055 | - | - | - | - |
| Pyrogallol | - | - | 0.0055 | - | - | - | - |
| Quercetin | - | - | 0.0018 | - | - | - | - |
| Resveratrol | - | - | 0.0018 | - | - | - | - |
| Sesamin | - | - | 0.0012 | - | - | - | - |
| Capsaicin | - | - | 0.0021 | - | - | - | - |
| Probucol | - | - | 0.00044 | - | - | - | - |
| Edaravon | - | - | 0.0067 | - | - | - | - |
* The 1O2 was generated in a dark reaction by thermodissociation from the respective endoperoxide. EDN, 1,4-dimethylnapthalene endoperoxide; NDPO2, of 3,3′-(1,4-naphthylene) dipropionate endoperoxide; EP, 1-methylnaphthalene-4-propionate endoperoxide; DPBF, 1,3-diphenylisobenzofuran; DMF, N,N-dimethylformamide; (−)-EGCG, (−)-epigallocatechin gallate.
 Carotenoids exhibit the necessary reactivity to function as antioxidants through favorable reactions with free radicals, including electron transfer and radical addition. AX and canthaxanthin demonstrate higher antioxidant effectiveness compared to β-carotene or zeaxanthin, despite slower rates of AMVN-induced oxidation. Resonance Raman spectroscopy and theoretical calculations are employed to investigate the molecular structures of radical species of AX, uncovering significant changes in bond orders and vibrational modes. The agreement between the theoretically calculated Raman spectra and the experimentally observed resonance Raman spectra enables a detailed discussion of the molecular structures of the radical species of AX, providing valuable insights into their functions in biological systems. Furthermore, although AX often forms fatty acid esters in nature, it has been reported that there is no essential difference in the first oxidation potential between AX and its n-octanoic mono- and diesters. This suggests that the AX esters have similar scavenging rates for •OH, •CH3, and •OOH radicals compared to AX itself [174][175].
Carotenoids exhibit the necessary reactivity to function as antioxidants through favorable reactions with free radicals, including electron transfer and radical addition. AX and canthaxanthin demonstrate higher antioxidant effectiveness compared to β-carotene or zeaxanthin, despite slower rates of AMVN-induced oxidation. Resonance Raman spectroscopy and theoretical calculations are employed to investigate the molecular structures of radical species of AX, uncovering significant changes in bond orders and vibrational modes. The agreement between the theoretically calculated Raman spectra and the experimentally observed resonance Raman spectra enables a detailed discussion of the molecular structures of the radical species of AX, providing valuable insights into their functions in biological systems. Furthermore, although AX often forms fatty acid esters in nature, it has been reported that there is no essential difference in the first oxidation potential between AX and its n-octanoic mono- and diesters. This suggests that the AX esters have similar scavenging rates for •OH, •CH3, and •OOH radicals compared to AX itself [174][175].
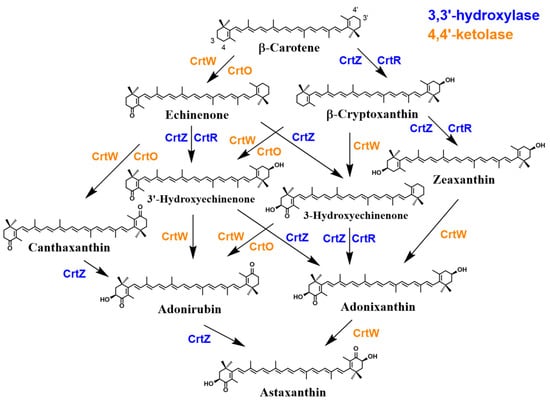 It was interestingly found that the phylum Cyanobacteria, which are photoautotrophic bacteria, possess a distinct β-C3-hydroxylase, designated CrtR, which shows moderate homology not to CrtZ but to CrtW [189]. Moreover, cyanobacteria were shown to retain a distinct β-C4-ketolase, designated CrtO, that shows significant homology to CrtI (phytoene desaturase) [190], in addition to CrtW. Thus, their presence could theoretically lead to the formation of AX. However, due to the substrate specificity of these enzymes, major carotenoids are not AX but its early-stage precursors such as 3′-OH-echinenone, echinenone, and zeaxanthin, as well as other cyanobacterium-related carotenoids such as myxol glycosides. Thus, AX is ordinarily not present or one of trace amounts of carotenoids in cyanobacteria [191][192][191,192], while previous reports, despite some debate regarding analytical methods and accuracy, described the presence of AX as a constitutive carotenoid in this phylum [193].
The reason is attributed to the extremely low or no reactivity of cyanobacterial CrtR towards a substrate with a 4-keto-β-end group. CrtR is ordinarily likely to mediate the synthesis of myxol 2′-fucoside by its β-C3-hydroxylase activity, while the Synechocystis sp. PCC 6803 CrtR can convert β-carotene into zeaxanthin, as shown by functional analysis using E. coli [189][191][189,191]. Similarly, functional analysis with E. coli demonstrated that cyanobacterial CrtO also exhibits very low or no reactivity towards a substrate with a 3-hydroxy-β-end group, since CrtO handles echinenone synthesis from β-carotene by its β-C4-ketolase activity (Figure 78) [194]. Moreover, it was found that cyanobacterial CrtW enzymes generally retain much lower activity for such a substrate, compared with Paracoccus and Brevundimonas CrtW proteins [195]. As a side note, a recent study reported that the introduction of the crtW and crtZ genes from Brevundimonas sp. SD212 into Synechococcus sp. PCC 7002, a type of cyanobacteria, resulted in the production and enhancement of AX productivity [191].
Although these AX products occur mainly in their free form in these bacteria, it has been reported that Agrobacterium aurantiacum (properly Paracoccus sp. strain N81106) produces a glycosylated AX, i.e., AX monoglucoside [196]. Additionally, Sphingomonas astaxanthinifaciens and S. lacus PB304 also contain AX dirhamnoside and AX dideoxyglucoside, respectively [197][198][199][197,198,199]. The enzyme gene that forms these glycosides, crtX, has been found in the complete gene clusters of Paracoccus sp. strain N81106 [37][200][37,200] and the genus Sphingomonas. Such an activity of CrtX was suggested using Pantoea ananatis CrtX with an AX-producing recombinant E. coli [201]. However, functional analysis using E. coli has not confirmed the ability of the putative gene of Sphingomonas to mediate such glycosylation reactions [188][198][188,198]. Furthermore, the presence of AX has also been implicated in the phyla Actinomycetota and Deinococcota (Deinococcus-Thermus); however, reliable structural analysis of AX and its biosynthetic genes in these phyla is still lacking. Further details regarding the bacterial AX biosynthetic pathway are discussed in Section 2.3.
It is indeed plausible that AX may play a protective role in cells exposed to intense sunlight and high levels of natural radiation. Bacteria that produce AX, such as those found in ocean surfaces, coastal areas, and hot springs, inhabit environments where they are subjected to these harsh conditions [181][182][183][184][196][202][181,182,183,184,196,202]. AX, with its antioxidant properties, has the potential to scavenge free radicals generated by UV radiation and protect cells from oxidative damage. Furthermore, AXs ability to absorb and dissipate excess light energy may also contribute to cellular photoprotection. These mechanisms suggest that AX could serve as a natural defense mechanism against the damaging effects of sunlight and radiation on these bacteria.
The presence of AX in halophilic archaea, which thrive in high-salinity environments where other organisms cannot survive, has been suggested [203][204][203,204]. In particular, studies on Halobacterium salinarum R1 have shown that depletion of the CYP174A1 gene, which codes for a cytochrome P450 (CYP; P450), resulted in decreased production of AX. It is noteworthy that the genome of H. salinarum does not contain genes encoding CrtZ-type or CrtR-type β-C3-hydroxylase, CrtW-type β-C4-ketolase, or CrtO-type β-C4-ketolase, which may be involved in AX production in other organisms [204]. This suggests that CYP174A1 may have a role in the biosynthesis of AX in these halophilic archaea. However, the presence and distribution of AX in archaea as a whole have yet to be fully confirmed, and further research is needed to elucidate this aspect.
It was interestingly found that the phylum Cyanobacteria, which are photoautotrophic bacteria, possess a distinct β-C3-hydroxylase, designated CrtR, which shows moderate homology not to CrtZ but to CrtW [189]. Moreover, cyanobacteria were shown to retain a distinct β-C4-ketolase, designated CrtO, that shows significant homology to CrtI (phytoene desaturase) [190], in addition to CrtW. Thus, their presence could theoretically lead to the formation of AX. However, due to the substrate specificity of these enzymes, major carotenoids are not AX but its early-stage precursors such as 3′-OH-echinenone, echinenone, and zeaxanthin, as well as other cyanobacterium-related carotenoids such as myxol glycosides. Thus, AX is ordinarily not present or one of trace amounts of carotenoids in cyanobacteria [191][192][191,192], while previous reports, despite some debate regarding analytical methods and accuracy, described the presence of AX as a constitutive carotenoid in this phylum [193].
The reason is attributed to the extremely low or no reactivity of cyanobacterial CrtR towards a substrate with a 4-keto-β-end group. CrtR is ordinarily likely to mediate the synthesis of myxol 2′-fucoside by its β-C3-hydroxylase activity, while the Synechocystis sp. PCC 6803 CrtR can convert β-carotene into zeaxanthin, as shown by functional analysis using E. coli [189][191][189,191]. Similarly, functional analysis with E. coli demonstrated that cyanobacterial CrtO also exhibits very low or no reactivity towards a substrate with a 3-hydroxy-β-end group, since CrtO handles echinenone synthesis from β-carotene by its β-C4-ketolase activity (Figure 78) [194]. Moreover, it was found that cyanobacterial CrtW enzymes generally retain much lower activity for such a substrate, compared with Paracoccus and Brevundimonas CrtW proteins [195]. As a side note, a recent study reported that the introduction of the crtW and crtZ genes from Brevundimonas sp. SD212 into Synechococcus sp. PCC 7002, a type of cyanobacteria, resulted in the production and enhancement of AX productivity [191].
Although these AX products occur mainly in their free form in these bacteria, it has been reported that Agrobacterium aurantiacum (properly Paracoccus sp. strain N81106) produces a glycosylated AX, i.e., AX monoglucoside [196]. Additionally, Sphingomonas astaxanthinifaciens and S. lacus PB304 also contain AX dirhamnoside and AX dideoxyglucoside, respectively [197][198][199][197,198,199]. The enzyme gene that forms these glycosides, crtX, has been found in the complete gene clusters of Paracoccus sp. strain N81106 [37][200][37,200] and the genus Sphingomonas. Such an activity of CrtX was suggested using Pantoea ananatis CrtX with an AX-producing recombinant E. coli [201]. However, functional analysis using E. coli has not confirmed the ability of the putative gene of Sphingomonas to mediate such glycosylation reactions [188][198][188,198]. Furthermore, the presence of AX has also been implicated in the phyla Actinomycetota and Deinococcota (Deinococcus-Thermus); however, reliable structural analysis of AX and its biosynthetic genes in these phyla is still lacking. Further details regarding the bacterial AX biosynthetic pathway are discussed in Section 2.3.
It is indeed plausible that AX may play a protective role in cells exposed to intense sunlight and high levels of natural radiation. Bacteria that produce AX, such as those found in ocean surfaces, coastal areas, and hot springs, inhabit environments where they are subjected to these harsh conditions [181][182][183][184][196][202][181,182,183,184,196,202]. AX, with its antioxidant properties, has the potential to scavenge free radicals generated by UV radiation and protect cells from oxidative damage. Furthermore, AXs ability to absorb and dissipate excess light energy may also contribute to cellular photoprotection. These mechanisms suggest that AX could serve as a natural defense mechanism against the damaging effects of sunlight and radiation on these bacteria.
The presence of AX in halophilic archaea, which thrive in high-salinity environments where other organisms cannot survive, has been suggested [203][204][203,204]. In particular, studies on Halobacterium salinarum R1 have shown that depletion of the CYP174A1 gene, which codes for a cytochrome P450 (CYP; P450), resulted in decreased production of AX. It is noteworthy that the genome of H. salinarum does not contain genes encoding CrtZ-type or CrtR-type β-C3-hydroxylase, CrtW-type β-C4-ketolase, or CrtO-type β-C4-ketolase, which may be involved in AX production in other organisms [204]. This suggests that CYP174A1 may have a role in the biosynthesis of AX in these halophilic archaea. However, the presence and distribution of AX in archaea as a whole have yet to be fully confirmed, and further research is needed to elucidate this aspect.
554]. Genetic and transcriptome analyses revealed that CYP2J2 and CYP2J6 were highly expressed in orange individuals compared to yellow individuals, suggesting that these P450s contribute to the conversion of carotenoids into ketocarotenoids [313].
Scavenging of Free Radicals and Inhibition of Lipid Peroxidation
Carotenoids can occasionally display pro-oxidant activity by oxidizing other lipids instead of acting as antioxidants, especially under high oxygen partial pressure or in the absence of other antioxidants. However, in comparison to other carotenoids, AX has shown minimal pro-oxidant activity in both simple solvent-based systems and evaluation systems that involve phospholipid membranes [142][159][143,160]. This is in contrast to lycopene, which is often assessed for its antioxidant properties. Studies employing pulse radiolysis and time-resolved spectroscopy have revealed that carotenoids engage with free radicals through different mechanisms [160][161][162][163][164][161,162,163,164,165]. The preliminary products formed during the interaction between carotenoids and free radicals indicate that electron transfer and radical addition are kinetically favored reactions. These reactions result in the oxidation of the carotenoid to its radical cation or the generation of carotenyl adduct radicals, such as [R-Car]• [165][166]. Carotenoids possess the necessary reactivity to function as antioxidants, and their reaction rates with free radicals are comparable to those of polyunsaturated fatty acids reacting with the same oxidants [142][143]. When comparing the reactivity of carotenoids with free radicals, it is observed that carotenoid radical cations can be reduced by α-, β-, and γ-tocopherol, while lycopene and β-carotene can reduce δ-tocopherol radicals [160][166][161,167]. β-Carotene•– transfers an electron to oxygen but not vice versa, while lycopene undergoes reversible electron transfer with O2•– due to its more positive electronegativity resulting from its planar geometry. Among the common carotenoids, AX radical cations are the most easily reduced. This implies that AX radical cations can be reduced by other carotenoids, like lycopene. Additionally, the interaction between carotenoids and other antioxidants may significantly contribute to their antioxidant activity in vivo. Skibsted and colleagues demonstrated that AX radical cation is effectively reduced by polyphenols such as isoflavonoids, in addition to the antioxidants mentioned earlier [167][168]. Interestingly, AX exhibits lower reducing abilities compared to other carotenoids. When reacting with CCl3OO•, AX does not directly form a radical cation but instead undergoes an addition of a radical, which subsequently decays to form the cation radical [164][165]. Despite this, AX and canthaxanthin demonstrate greater effectiveness as antioxidants compared to β-carotene or zeaxanthin in retarding hydroperoxide formation during azo-initiated lipid peroxidation in homogeneous methyl linoleate/AMVN systems. However, the rates of AMVN-induced oxidation of AX and canthaxanthin are slower than those of β-carotene and zeaxanthin [168][169]. These findings indicate that AX and other carotenoids, when consumed in combination with multiple antioxidants rather than individually, may have a better ability to inhibit lipid peroxidation through the interaction of antioxidant networks. This could provide an explanation for the suggestion that aggressive supplementation of synthetic β-carotene at high doses may actually increase the risk of lung cancer in smokers and asbestos-exposed workers [169][170][170,171]. On the other hand, simultaneous intake of green and yellow vegetables, which contain multiple carotenoids and antioxidant vitamins, has been associated with a reduced risk of cancer [171][172][173][172,173,174]. Therefore, understanding the reactivity of AX with reactive oxygen species (ROS) and free radicals, as well as the physicochemical properties of its reaction intermediates, is crucial for a comprehensive understanding of the true antioxidant activity of AX.Structures of Radical Cation and Dication of Astaxanthin as Predicted Based on DFT Calculations and Resonance Raman Spectroscopy
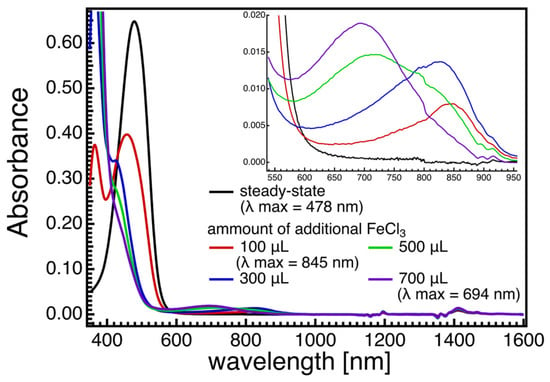
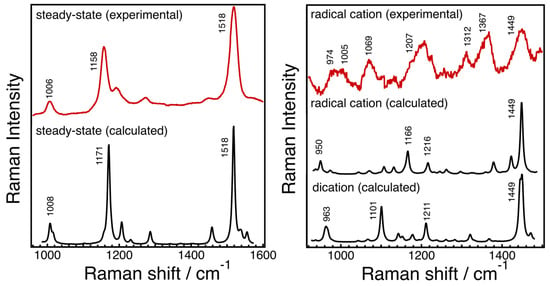
The results from pulse radiolysis and time-resolved spectroscopy studies demonstrate that carotenoids possess the necessary reactivity to function as antioxidants through kinetically favored reactions, including electron transfer and radical addition when interacting with free radicals. Various tocopherols can reduce carotenoid radical cations, and lycopene can undergo reversible electron transfer with O2•– due to its positive electronegativity. AX and canthaxanthin exhibit higher antioxidant effectiveness compared to β-carotene or zeaxanthin, despite slower rates of AMVN-induced oxidation. Resonance Raman spectroscopy, along with theoretical calculations, has been utilized to investigate the molecular structures of radical species of AX, revealing significant changes in bond orders and vibrational modes. Figure 45 displays the steady-state absorption spectra of AX in acetone with different amounts of FeCl3 solutions (1 mM acetone solution) added. The addition of FeCl3 solution oxidizes AX, resulting in the formation of a radical cation peaking around 850 nm and a dication peaking around 700 nm, depending on the amount of FeCl3 solution added. Resonance Raman spectra of AX and its radical species were recorded and compared with the DFT calculations in Figure 56. The DFT calculations accurately replicate the ground-state (S0) Raman spectrum, while the resonance Raman spectrum of the radical species can be seen as a combination of the calculated Raman spectra of the radical cation and the dication of AX. This is due to the fact that the resonance Raman spectrum of the ground (S0) state species was recorded using 532 nm laser light, which resonates with the S0 → S2 absorption of AX, while the resonance Raman spectrum of the radical species was recorded using 808 nm laser light, which is in resonance with both the radical cation and dication of AX. The agreement between the theoretically calculated Raman spectra and the experimentally observed resonance Raman spectra enables a detailed discussion of the molecular structures of the radical species of AX. As an example, Figure 67 illustrates the bond lengths of the ground (S0) state, radical cation, and dication of AX. It is noteworthy that the bond alterations evident in the ground (S0) state species undergo dramatic changes in the radical cation species, with all C=C double bonds tending to elongate and all C-C single bonds tending to shrink. This suggests that the bond orders of all the C=C and C-C bonds in the conjugated polyene approach 1.5. In the case of the dication species, there is a striking reversal of bond alterations in the central part of the polyene chain, with C=C double bonds becoming C-C single bonds and C-C single bonds becoming C=C bonds. These significant changes in bond orders are accurately reflected in the vibrational modes, which were predicted based on DFT calculations. The theoretically predicted molecular structures of the radical species of AX, supported experimentally through resonance Raman spectroscopy, serve as a valuable tool for exploring the functions of the radical species of AX in biological systems.Figure 45. Steady-state absorption spectra of astaxanthin (AX) in acetone when different amounts of FeCl3 solutions (1 mM acetone solution) were added. According to the addition of the FeCl3 solution, the intensity of S0 → S2 absorption of AX around 500 nm decreases, and the new absorption bands appear in the 600–950 nm spectral region (see inset of Figure 45). With the small amount of FeCl3 solution added, the absorption band that is associable to the radical cation of AX appears to peak around 850 nm. With more addition of the FeCl3 solution, the radical cation of AX transforms to dication, peaking around 700 nm. The absorption band below 400 nm is due to the absorption of FeCl3.
Figure 56. The steady-state resonance Raman spectrum of astaxanthin (AX) in acetone recorded with 532 nm excitation laser light at room temperature (solid red line in the left panel) and the resonance Raman spectra of radical species of AX recorded with 808 nm excitation laser light at room temperature (solid red line in the right panel). The results of DFT calculations of the ground (S0) species, radical cation, and dication of AX are also shown in each panel (solid black lines).
Figure 67. Comparison of the bond lengths of the ground (S0) state, radical cation, and dication of astaxanthin predicted theoretically by DFT calculations.
Biochemical Aspects of AX Properties against ROS
The intracellular localization and ROS scavenging activity of AX were presented in an earlier review [175][130]. To summarize, inflammation, whether acute or chronic, generates ROS and often leads to oxidative stress in vivo. Important actions of AX include the inhibition of nuclear translocation of NFκB, which promotes inflammatory responses, and the activation of Nrf2, a transcription factor of a group of anti-inflammatory enzymes. In parallel, oxidative stress caused by ROS can be reduced by improving mitochondrial function, which is a major source of ROS in vivo [175][176][130,176]. In conclusion, AX could exert its typical effects in vivo by inducing multifaceted antioxidant activity beyond the antioxidant activity derived from the chemical properties of the compound itself. Their typical efficacies are shown in Section 3.2.2. Astaxanthin; Distribution, Derivatives and Optical Structure in Nature
AX is found in a considerable number of organism species, which cover a wide taxonomic variety that ranges from bacteria to several eukaryote kingdoms, i.e., fungi (yeasts), algae, higher plants, and animals. Table 2 provides information on the approximate amounts and isomers of AX in major species. With the exception of animals, organisms that possess AX are capable of synthesizing it de novo through either the non-mevalonate pathway (also known as the MEP pathway) or the mevalonate pathway. The non-mevalonate pathway involves the conversion of pyruvate and glyceraldehyde 3-phosphate into 1-deoxy-D-xylulose-5-phosphate (DXP) and 2-C-methyl-D-erythritol-4-phosphate (MEP) [177]. Further details on these pathways can be found in Section 2Section 2.3.3.2.2.1. Bacteria and Archaea
Among bacteria, the genus Paracoccus, belonging to the class α-Proteobacteria (Alphaproteobacteria) in the phylum Proteobacteria, has been found to produce (3S,3′S)-AX [178][179][180][178,179,180]. Additionally, other bacteria such as Brevundimonas, Sphingomonas, and Altererythrobacter species have also been reported to have the ability to produce AX [181][182][183][184][185][186][181,182,183,184,185,186]. In these bacteria, AX biosynthesis occurs from β-carotene through oxygenation reactions catalyzed by two enzymes called CrtZ and CrtW. The two enzymes are β-C3-hydroxylase [β-carotene (β-carotenoid) 3,3′-hydroxylase] and β-C4-ketolase [β-carotene (β-carotenoid) 4,4′-ketolase (oxygenase)], and facilitate multistep hydroxylation and ketolation (oxygenation) reactions at the C3 and C4 positions of the β-ionone ring (β-end group), respectively (Figure 78) [37]. The CrtZ-type β-C3-hydroxylase responsible for the hydroxylation of the C3 and C3′ positions of the β-ionone rings generates a single stereo configuration, resulting in the production of (3S,3′S)-AX with a specific absolute optical configuration [187][188][187,188].Figure 78. Biosynthetic pathway of astaxanthin from β-carotene with bacterial enzymes. β-Carotene (β-carotenoid) 3,3′-hydroxylase and 4,4′-ketolase are shown with blue and orange letters, respectively. In this figure, the maximal levels of catalytic activities are shown concerning CrtR and CrtO. Generally, the catalytic activity from adonixanthin to astaxanthin is weak, even with CrtW. This pathway is based on bacterial enzymes. However, the functions of green algal BHY and BKT are the same as those of CrtZ and CrtW, respectively.
2.2.2. Eukaryotes; Fungi and Protozoa
Certain colored fungi are capable of producing carotenoids, including xanthophylls [205]. However, the production of AX (astaxanthin) in fungi is quite limited and is currently only observed in the genus Xanthophyllomyces. The yeast Phaffia rhodozyma, which is the anamorph of Xanthophyllomyces dendrorhous, was initially isolated from exudates of deciduous broadleaf trees at high altitudes in the northern hemisphere, where it is exposed to intense UV radiation [206][207][206,207]. In recent years, similar species belonging to the genus Xanthophyllomyces have also been discovered in the southern hemisphere, such as in Patagonia, South Australia, and Tasmania [208][209][210][208,209,210]. The differences in host trees and the evolutionary distances between these Xanthophyllomyces species from the northern and southern hemispheres suggest that they have been strongly influenced by the ancient continental separation of the Earth, which is of great interest from an Earth science perspective [211]. Phaffia yeast, including Xanthophyllomyces species, produce (3R,3′R)-AX in its free form [51]. The biosynthesis pathway of AX in Phaffia yeast is catalyzed by specific monooxygenase enzymes belonging to the P450 family, such as CrtS/Asy, along with a reductase enzyme. This pathway differs significantly from the metabolic pathway observed in bacteria [212][213][212,213]. It is believed that AX plays a crucial cytoprotective role for Phaffia yeast in adapting to these harsh environments. The biosynthesis of AX in Phaffia yeast is likely induced by redox imbalances, particularly those involving the NAD(P)H/NAD(P)+ couple and the oxidative environment [214]. Overall, the production of AX in fungi, particularly in Phaffia yeast, is an intriguing phenomenon, and its biosynthesis pathway and protective role in challenging environments are subjects of scientific interest and investigation. Several zooplankton and phytoplankton species have the ability to synthesize AX de novo. The genera Nannochloropsis in the class Eustigmatophyceae, Aurantiochytrium in the class Labyrinthulea, and Euglena and Trachelomonas volvocina in the class Euglenophyceae, as well as certain arthropods like copepods and krill, have been reported to have the potential for AX production [215][216][217][218][219][215,216,217,218,219] (Table 2). These findings suggest the possibility of industrial-scale production of AX from these organisms. With advances in biotechnology and cultivation techniques, there is increasing interest in harnessing the AX production capacity of these zooplankton and phytoplankton species for commercial purposes. Industrial production of AX from these natural sources could provide a sustainable and renewable supply of this valuable compound for various applications. However, further research and optimization are required to fully exploit their potential and scale up the production process effectively.2.2.3. Eukaryotes; Algae and Higher Plants
It is worth noting that several green algae are known to accumulate extremely high concentrations of AX in cells under high sunlight, high salinity, and starvation conditions. The most well-known example is the green algae of the genus Hemacotococcus, particularly H. lacustris (Gir.-Chantr.) Rostaf. (=H. pluvialis Flot.). Please note that this document supports the official scientific name of H. lacustris (Gir.-Chantr.) Rostaf., as per the comprehensive opinion of Ota et al. [220][221][220,221]. Descriptions of Haematococcus algae (=H. lacustris), its biology, and life cycles emerged from the works of the German botanist Julius von Flotow in 1844 and the American botanist Tracy Elliot Hazen in 1899. Although early botanists described a “blood red pigment” produced by this algae [222], it was later determined that this was AX [49][223][49,223]. Nevertheless, since the description of “H. pluvialis Flot.” can also be frequently found in numerous publications, this species is referred to as “Haematococcus algae” in this paper to avoid misunderstandings, unless there are exceptions in specifying the strain name. Recent molecular genetics re-classification has revealed that H. lacustris strains are highly genetically diverse [224], and other species previously assigned to the Haematococcus genus may be reclassified as a separate genus, possibly leaving only one species in this genus, H. lacustris [48]. The properties and industrial production of this alga are presented in detail in Section 3Section 3.3.3 of original paper. In Haematococcus algae, (3S,3′S)-AX is present in the form of a fatty acid ester bonded to the hydroxyl group at the C3, C’3 position, with the main component being a monoester. Other specific species of green algae belonging to the genera Acutodesmus, Asterarcys, Bracteacoccus, Botryococcus, Chlamydomonas, Chlorella (Chromochloris), Chlorococcum, Coelastrella, Monoraphidium, Neochloris, Protosiphon, Sanguina, Scenedesmus, Scotiellopsis, Tetraedron, and Vischeria have also been reported to accumulate AX mainly in esters and/or free form, as well as in protein binding forms [114][225][226][227][228][229][230][231][232][233][234][235][236][237][238][239][240][241][242][114,225,226,227,228,229,230,231,232,233,234,235,236,237,238,239,240,241,242]. Another example is the fatty acid esters of AX diglucoside, which have been reported from a low-temperature-tolerant algae (Chlamydomonas nivalis) that grows on snowfields and glaciers in the Alps and polar regions worldwide [229]. Interestingly, a very recent report isolated Dysmorphococcus globosus-HI, belonging to the family Phacotaceae within the order Chlamydomonadales, class Chlorophyceae, from the Himalayan region of northern India, and it was found that AX accounts for about 56% of the dry intracellular weight. However, reproducibility and further studies on this report seem to be needed [243]. Additionally, many other phytoplankton also produce or accumulate carotenoids, including AX [244][245][244,245]. Therefore, it is likely that the accumulation of carotenoids in higher animals through the food chain is a result of the presence of carotenoids in plankton [246]. In these algae, AX provides three biological advantages to the algal cells: (1) stabilization of quiescent cells. Some of these algae form non-motile, extra-long-life, environmentally resistant cysts called aplanospores under severe environmental stress, as described above. These cysts are quiescent cells similar to plant seeds, with decreased chloroplast volume and a high accumulation of oil droplets containing high concentrations of AX in the cytoplasm [223][247][248][249][250][251][223,247,248,249,250,251]. In the case of Haematococcus algae, mature cyst cells (also often described as aplanospores [221][252][221,252] or akinetes [224]) exhibit a deep scarlet color. The biological mechanism of this unique accumulation in oil droplets is shown in detail in Section 2.3.1, which provides an example of the biosynthesis of AX in Haematococcus algae. AX is believed to protect the cells and their stored components, such as lipids and DNA, from the surrounding severe environment, including intense light exposure [253]. (2) In snow algae, AX surrounds and masks the chloroplasts in the algae, thereby promoting the ability to utilize red light wavelengths, which are much more effective for CO2 uptake than green or blue light [254]. (3) Similar to the putative role in (2), it has been reported that in Haematococcus algae, AX in green-red non-motile cyst cells, often called palmelloids before becoming scarlet-colored mature quiescent cyst cells, is localized in the cell center (the nuclear periphery) under normal light conditions but rapidly diffuses to the cell periphery when exposed to strong light. In cells where AX has completely moved to the periphery, a thin layer of AX is identified just beneath the cell wall, and if these cells are placed in the dark, AX redistributes to the center of the cell (the nuclear periphery) [250][255][250,255]. It has been hypothesized that this translocation may be facilitated by actin filaments [255]. In all cases (1) to (3), AX likely performs a similar physiological function in terms of protecting photosynthetic organs and intracellular components from intense light. Other organisms, such as E. sanguinea, which accumulates AX fatty acid esters [217][256][217,256], and C. astaxanthina Ki-4, which produces a complex of AstaP and AX [257], s show similar responses that accumulate AX derivatives under intense light, suggesting that AX plays the same role in a wide variety of microalgal species. In terrestrial higher plants, carotenoids serve as accessory pigments in photosynthesis. They capture light energy as antennae pigments and also quench reactive oxygen species (ROS) generated by photosynthetic reaction centers under intense light [112] (see also Section 2.1.7). These primary carotenoids are typically located in the thylakoid membranes of chloroplasts and are essential for efficient photosynthetic reactions. They are biosynthesized in chloroplasts and are present within chloroplasts as well [258][259][258,259]. However, terrestrial higher plants often accumulate carotenoids in chromoplasts or lipid globules that are not directly involved in photosynthesis. Examples include the carotenoids found in fruits, flowers, and roots of many plants, such as lycopene in tomatoes and β-carotene in carrot roots. These carotenoids are known as secondary carotenoids [5][259][5,259]. AX is rarely found as a major carotenoid involved in photosynthetic function in higher plants, as mentioned previously. Therefore, AX is considered a secondary carotenoid rather than a primary carotenoid essential for photosynthesis. Some earlier studies reported the presence of AX in specific higher plants [260]. However, it was later confirmed that the reddish carotenoids found in autumn leaves were actually rhodoxanthins and their metabolites [261][262][261,262]. Currently, the only terrestrial plants where AX has been reliably identified as a major carotenoid are the reddish petals of certain species belonging to the genus Adonis of the Ranunculaceae family. In these plants, (3S,3′S)-AX is present in the form of fatty acid diesters [17][263][264][265][17,263,264,265]. Nevertheless, there remains a possibility that higher plants containing AX will be discovered in the future, as undiscovered plant species may still exist worldwide.2.2.4. Eukaryotes; Animals
With a few exceptions in certain arthropods, all animals lack the ability to produce carotenoids de novo [266]. Therefore, the AX that exists is either obtained from the diet or metabolically converted from precursor carotenoids. The coloration of invertebrates and poikilothermic vertebrates primarily depends on the types of chromatophores present in their integuments (i.e., skin and exoskeleton) (see Supplementary Table S3 of original paperSupplementary Table S3). While there are various types of chromatophores, both carotenoids and phenolic pteridines contribute to the red and yellow color patterns. Specifically, red chromatophores, known as erythrophores, contain ketocarotenoids like AX. Research on animal coloration, focusing on pigments, has predominantly examined ornamental color signals that indicate animal maturity and provide reproductive advantages. Recent hypotheses propose that oxidative stress plays a crucial role in maintaining the honesty of condition-dependent carotenoid-based signaling [267][268][269][267,268,269]. This is due to the potential of carotenoids to serve as honest indicators of phenotypic quality and, consequently, as targets for resource allocation trade-off hypotheses. In many animals, the amount of pigment available in vivo is limited, and its allocation may involve a trade-off between the animal’s mature phenotype and various essential biological functions, such as the immune system and antioxidant defense [270][271][270,271]. Concurrently, pigments have been extensively linked to prey-predator relationships [272][273][274][275][272,273,274,275]. Numerous studies have demonstrated that prey species exhibit vibrant colors as a means to signal the presence of predator defenses, thus serving as a mechanism to deter predators. In other words, predators advertise their defenses by being conspicuous, which enhances predator recognition and avoidance learning through uniform signaling. Long-wavelength color patches, such as red, orange, and yellow, are recognized as effective components of many visual warning signals, particularly when combined with black [276]. The red color derived from ketocarotenoids, including AX, holds significant prominence, especially for mammals like us.In Case of Invertebrates
As mentioned above, AX contributes to the colorful body color and internal organs of invertebrates. Information on the distribution and conversion of carotenoids in aquatic organisms is detailed in Matsuno’s comprehensive review [21]. Similarly, another review on stereoisomers of hydroxyl groups at the C3 and C’3 positions of AX in aquatic animals is detailed in Matsuno et al. [277]. In luminal animals, AX and “nor”-carotenoids, such as 2-norastaxanthin and actinioerythrin, are present in sea anemones [278], where the carbon at the 2 or 2′ position of AX is detached. AX has also been reported in jellyfish and corals [279][280][279,280]. It is taken up from crustacean-like zooplankton as a dietary source [280][281][280,281]. In mollusks, (3S,3′S)-AX has been found in coiled mollusks such as the spindle snail (Fusinus perplexus) and the apple snail (Pomacea canaliculata) [21]. Triton’s trumpet also contains AX, which is produced through the oxidative metabolic conversion of β-carotene and (3R,3′R)-zeaxanthin from starfish [281]. It is a mixture of three optical isomers that are taken up by the starfish when they feed on it. In bivalves, (3S,3′S)-AX is present as a trace component along with pectenolone in the ovaries of the Yesso scallop (Mizuhopecten yessoensis) [281][282][281,282]. Additionally, some Yesso scallops have orange adductor muscles [283]. Moreover, various colorful shells are found in the noble scallop (Mimachlamys crassicostata, synonym: Chlamys nobilis), with some individuals having golden shells and scallops with golden inner tissues [284]. These individuals have a high accumulation of carotenoids. The reasons for the difference in coloration of these bivalves are discussed in Section 2.3Section 2.3.3.3. Interestingly, it has been reported that these golden individuals have higher expression of antioxidant enzymes and improved immunological responses, as well as increased resistance to low-temperature stress compared to their brown counterparts [285][286][287][285,286,287]. AX is present in the viscera and ovaries of cephalopods (squids and octopuses) as a mixture of three stereoisomers [288]. In arthropods, the optical isomer composition of AX can be divided into three groups: (1) consisting of only one isomer or more than 90% of the dominant isomer; (2) mostly containing two isomers with a trace amount or a small proportion of another isomer; and (3) a mixture of all three isomers, with two at similar levels and the third in larger or smaller quantities (Table 2). While the majority of arthropods rely on dietary carotenoids, certain crustaceans, such as shrimps, prawns, and crabs, can convert β-carotene into AX through oxidative reactions involving echinenone, canthaxanthin, and adonirubin. With the exception of group (1), the AX in these cases consists of a mixture of three optical isomers due to the likely lack of stereospecificity in introducing hydroxyl groups at the C3 and C3′ positions [281]. In other arthropods, the conversion to AX appears to utilize β-carotene and zeaxanthin as substrates. Acari and copepods predominantly produce (3S,3′S)-AXs through this process [289][290][289,290]. However, it is noteworthy that Antarctic krill exhibits significantly higher levels of AXs with the (3R,3′R) stereo configuration [277][291][277,291]. Further details are provided in Section 2.3.3. Additionally, these AXs can exist in free, monoester, and diester forms. Furthermore, some AXs are present as protein complexes called crustacyanins [83]. Detailed information on crustacyanins was provided in Section 2Section 2.1.5.1.5. In lobsters (Homarus gammarus), crustacyanins, including β-crustacyanin (blue) and its octamer, α-crustacyanin (purple), and crustochrin (a yellow analog of these proteins), are found in the exoskeleton in the form of free AX associated stoichiometrically with apoproteins. Although the exoskeletons of these organisms are covered by a thick cuticular layer of epidermis, the localization of these characteristic carotenoproteins differs. In a study involving whitened American lobsters (H. americanus) fed a carotenoid-deficient diet, the accumulation of AX esters initially occurs in the epidermis, which then migrates and accumulates crustacyanins, resulting in the formation of a thick cuticle. Finally, multiple crustacyanin molecules are stacked like plates within the epicuticular layer to form crustochrin [292]. This study also demonstrated that UV irradiation promotes the accumulation of these carotenoproteins in the exoskeleton, suggesting that these pigments may serve as either cryptic coloration or protection against UV radiation [292]. On the outer surface of the carapace, where crustochrin is present, AX exists as an H1-aggregate, exhibiting a typical hypsochromic shift and absorbing light primarily in the UV region. Therefore, crustochrin itself exhibits a UV-shielding effect. Furthermore, the H1-aggregate of AX is considered less reactive with other compounds since there is no intermediate state for UV-induced triplet formation, and singlet fission occurs directly from the photoexcited state of 1Bu with an ultrafast time scale, which may be beneficial for the survival of lobsters from a physicochemical perspective [79] (see Section 2.1.6). An immunohistological study conducted on rock lobsters (Panulirus cygnus) also revealed the presence of cluster cyanine subunits not only in the cuticle layer of the exoskeleton but also in the epithelial layer. Moreover, P. cygnus is characterized by various patterned areas on its shell, including fine red and white spots and horizontal white stripes along the length of the shell. Immunohistochemical staining showed that crustacyanins were restricted to the colored areas on the carapace and the corresponding epidermis. These results suggest that the shell color pattern is formed through the expression of crustacyanins in the epithelium and their incorporation into the exoskeleton, rather than solely the absence of carotenoid chromophores [86]. In a study of the characterization and metabolism of carotenoproteins during embryonic development of the lobster H. gammarus, larvae contained carotenoproteins of three unknown structures (blue, red, and yellow) that were thought to be metabolized and transferred by an unknown factor (referred to as “AX”) from ovoverdin, the major carotenoprotein in egg yolk. Additionally, the egg yolk carotenoids were identified as free AX and adonirubin, while during tissue formation, AX was esterified. Therefore, the diester of AX was found to be the major carotenoid in embryos and larval tissues. This AX diester was found to be bound to red carotene proteins. The increase in esterified AX suggests that an early enzymatic mechanism leading to the acylation reaction occurs during embryogenesis [90]. AX has also been reported in insects, including grasshoppers [16][293][294][16,293,294] (Table 2). In most cases, these AX compounds are derived from their diet within the food chain. However, there are a few exceptions among arthropods, such as spider mites (Acari; e.g., Tetranychus urticae) [295][296][295,296] and aphids [297][298][299][297,298,299], which have the ability to synthesize carotenoids de novo. These organisms possess orthologous genes for enzyme proteins, namely CrtYB and CrtI, which are involved in the carotenoid biosynthetic pathway. It is believed that these genes originated in fungi and were acquired through lateral gene transfer [266][296][300][266,296,300] (for more details, refer to Section 2.3.3). In echinoderms, it has been reported that AX exists as a mixture of three stereoisomers in starfish, sea cucumbers, and sea urchins [301][302][303][301,302,303]. From starfish, AX and its triple-bonded derivatives, namely 7,8-didehydroastaxanthin and 7,8,7′,8′-tetradehydroastaxanthin, have been identified [280][304][280,304]. Previously, 7,8-didehydroastaxanthin and 7,8,7′,8′-tetradehydroastaxanthin were referred to as asterinic acid, named after the scientific name of the starfish species (Asterias rubens) [304][305][304,305]. In protochordates, the presence of (3S,3′S)-AX has been reported in sea squirts (Halocynthia roretzi), along with other major carotenoids [306].In Case of Vertebrates
In fish, the presence of AX depends on the food chain, which means it is the result of absorption, accumulation, or metabolism from the diet. The red color on the body surface of many white meat fish species, apart from red meat fish, is attributed to AX and serves as the basis for the characteristic body color of each species. The origin of these body colors can be divided into the following patterns: (1) Salmonidae, including salmon and trout, which contain AX in their skeletal muscles; and marine white meat fishes with a red body surface, such as red snapper; and (2) carp fishes, which can metabolically convert AX from zeaxanthin. The former accumulates AX derived from crustaceans, which are their major food source, resulting in a mixture of three stereoisomers of AX [281]. The latter metabolically converts (3R,3′R)-zeaxanthin as a precursor, leading to the presence of only (3S,3′S)-AX [24][307][24,307]. Due to these characteristics, AX is often used as a coloring agent for snapper and salmon, while zeaxanthin is used as a coloring agent for goldfish (colored varieties of Carassius auratus) and Nishikigoi (colored varieties of Cyprinus carpio) [307]. Moreover, marine fish are frequently fed zeaxanthin as a coloring agent. The metabolism and conversion to and from AX will be discussed in detail in Section 2Section 2.3.3.3.3. In the case of salmonid fish, AX is believed to originate from minuscule arthropods referred to as zooplankton. These zooplankton serve as prey for salmonid fishes, which in turn accumulate the pigments derived from them (refer to Table 2). Furthermore, there are significant differences in carotenoid composition between land-locked and migratory forms of salmonids due to variations in their dietary habits. Among salmonid fishes, the highest concentrations of AX have been reported in Sockeye salmon, and the ratio of AX to total carotenoids is also high in their muscles and eggs [308] (Table 2). Additionally, numerous fish species, particularly those belonging to the red-fleshed and salmonid categories, undergo metabolic conversion of AX into yellow xanthophylls like tunaxanthin or salmoxanthin via a reductive pathway. These yellow pigments are then deposited and stored in their integument (skin). Further elaboration on these processes will be provided in the forthcoming Section 2Section 2.3.3.3.3. In amphibians, the presence of AX has been reported in newts and frogs/toads. In these organisms, AX exists in both free and ester forms (Table 2). It is believed that AX, along with other carotenoids, plays a significant role in warning coloration against predators and in the nuptial coloration that indicates individual maturity. Carotenoids responsible for body coloration are thought to be present in the dermis as xanthophores and erythrophores, as well as in liver tissue [309]. However, their specific forms in other tissues remain unclear. Similar to red-skinned fish, AX is believed to be present in erythrospores found in integument tissues. Since many amphibians feed on insects, it is possible that some carotenoids derived from arthropods, mainly carotenes, accumulate intact or undergo oxidative conversion to become AX. Unfortunately, many of the studies on this topic are relatively old and were conducted before the prevalence of HPLC. With a few exceptions, such as the Japanese newt studies, they provide limited information [309][310][309,310]. Therefore, further instrumental analysis is required to determine the precise form in which AX exists. Recently, it was reported that certain frogs may possess enzymatic genes that facilitate the conversion of C4-ketocarotenoids, such as AX, through oxidative metabolism [294] (for more details, refer to Section 2.3.3). In reptiles, certain lizards and snakes display vibrant ornamental colors on their skin as a sign of maturity. These colors often include shades of yellow and red, which are attributed to the accumulation of high concentrations of carotenoids and phenolic pigments, such as pteridines. These orange to red pigments are typically found in the form of ketocarotenoids and, in some cases, contain high levels of AX (Table 2). In reptiles, these ketocarotenoids accumulate in the dermis within erythrophores [311][312][313][311,312,313]. Similar to reptiles, avian species also exhibit red-orange ornamental colors derived from C4-ketocarotenoids like AX [314]. In birds, AX has been reported in numerous species [314], most notably contributing to the body coloration of flamingos [15]. Interestingly, the ratio of AX to total carotenoids in flamingo feathers is relatively low, with canthaxanthin being the most abundant ketocarotenoid in many species. However, all species still contain significant amounts of AX, particularly in the legs of American flamingos [315]. While in most bird species, AX is visually identifiable in feathers, beaks, and legs, it has also been found in the skin beneath the plumage and other body tissues (e.g., liver, blood, etc.). As with other animals, there exists a trade-off between carotenoid ornamentation of the body surface and other physiological functions due to the limited dietary supply of carotenoids. These ketocarotenoids were initially believed to indicate individual maturity and be involved in defense against oxidative stress and regulation of immunity. However, the results have been inconsistent, and meta-analysis suggests that their presence reflects individual quality rather than consistently reducing oxidative stress [269]. The metabolism of these pigments is discussed in Section 2Section 2.3.3.3.3. Considering the localization of AX in avian tissues, it is believed to coexist with collagen matrices and lipids in the subcutaneous tissue, dermis, and epidermis. Unlike reptiles, birds lack specific chromatophores other than melanocytes in their skin tissue, including erythrophores. In feathers, AX is presumed to be present within the keratin matrices. Interestingly, AX is also found in the retinas of various bird species and turtles. It has been hypothesized that AX, along with other xanthophylls, may serve to protect retinal cells, particularly longwave-sensitive (LWS) cone cells, by acting as cut-off filters against blue light, especially in situations with intense light exposure [316][317][318][316,317,318]. Birds exhibit well-defined cone cells in the retina with oil droplets that contain specific carotenoids, including AX (R-type). These carotenoids are believed to have a wavelength-cutting effect on light, enhancing sensitivity in their respective wavelength ranges [316][317][318][316,317,318]. In LWS cone cells, the dominant optical isomers of AX are (3S,3′S)-AX [319]. In the majority of bird species, AX and other ketocarotenoids are presumed to be present in the retina, even in those without AX pigmentation in their plumage, with a few exceptions found in phylogenetic groups adapted to low-light environments, such as penguins and owls [320][321][322][320,321,322]. Penguins and owls inhabit nocturnal or deep-diving habitats and may have evolved colorless cone oil droplets to avoid interference with their vision, as colored droplets could impact their visual perception [318]. The functional role and evolutionary significance of these cone oil droplets are discussed in greater detail in other reviews (refer to [318][323][318,323]). In addition to color patterns, some animals utilize polarized light patterns as a means of communication. The biological polarizers found in nature rely on physical interactions with light, including birefringence, differential reflection, and scattering. Interestingly, among invertebrates, a species called the marine stomatopod crustacean, specifically the mantis shrimp (Odontodactylus scyllarus), possesses a unique biological polarizer based on the linear dichroism of carotenoid molecules in its antennal scale. These creatures are believed to have a fundamentally different color recognition system from ours and to perceive and experience the world of colors in a distinct manner [324]. In the mantis shrimp, AX is deposited on the antennal scales, and the presence of AX in this dichroic compound allows the scales to polarize light. Within these antennal scales, AX exists as an optical mixture. By utilizing AX as a dichroic material in their antennal scales, mantis shrimps are able to manipulate the polarization component of light rather than its color, generating signals that vary with the direction of polarization [325]. Hence, AX plays a crucial role in the visual function of diverse organisms, including those employing polarized light communication. In mammals, studies analyzing blood carotenoids have demonstrated that humans are capable of absorbing AX. Additionally, mice and rats have been observed to transfer AX to various tissues, including adipose tissue, liver, skeletal muscle, brain, and others [250][326][250,326]. More extensive information on these findings can be found in Section 4.1Section 4.1.3.3 of original paper. To summarize, animals discussed in SectionSection 2.2.3 2.2.3 and SectionSection 2.2.4 2.2.4 accumulate AX on their body surfaces, utilizing it for breeding purposes or as cryptic/warning coloration. AX is also found in abundance in eggs. It is speculated that AX may offer protection to eggs against light and reactive oxygen species (ROS) and is believed to be involved in reproductive functions, hatching, post-hatching survival, and growth. Further details and hypotheses regarding these aspects can be found in Section 3 and Section 5 of original paperSection 5.2.2.5. Astaxanthin Content in Various Organisms
The content of AX per 100 g in various organisms is as follows: lobster/crayfish—0.1–0.3 mg in the whole body, Sockeye salmon muscle and eggs—2.0–3.8 mg, krill—2.0–4.0 mg, Phaffia yeast—20–1000 mg [327], dry biomass of Paracoccus—20–2000 mg, and mature cyst cells of Haematococcus algae—up to 9800 mg [328][329][328,329] (Table 2). Among the reported species, Haematococcus algae has the highest AX content by a significant margin. Additionally, the purity of AX as a carotenoid is also high in Haematococcus algae. Therefore, the current industrial production of AX, apart from chemical synthesis, primarily relies on Haematococcus algae. As a result, most of the evidence supporting the use of AX as a dietary supplement for humans is based on Hematococcus algae. Other sources of AX, whether biological or synthetic, have limited applications in human consumption but are widely used in animal feed. Additionally, some products, such as krill oil, are valued for their pharmaceutical benefits derived from other components, like omega-3 fatty acids, rather than solely relying on the effects of AX. Consequently, here, the focus on human use of AX will primarily revolve around Hematococcus algae.Table 2.
Astaxanthin: Summary of its presence in various organisms and its origin.
| Taxon | Scientific Name | Common Name | Astaxanthin | Reference | |||
|---|---|---|---|---|---|---|---|
| Form † | Stereoisomer (3R,3′R, meso, 3S,3′ | ||||||
Table 3.
Summary of P450 enzymes with possible C4 ketolase activity using zeaxanthin or β-carotene as substrates.
| Name P450 |
Origin | Super-Family, Clan | Methodlogy of Functional Analysis | Reference | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| S | ) | Content (mg/100 g) | Origin | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Bacteria, Prtoteobacteria, Alphaproteobacteria | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| CYP2J19 | Aves/Testudines | CYP2 | Genetics Heterologous expression Homology |
[321]][321[552],552[553][557,553,557] | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Paracoccus carotinifaciens (w/. mutation) |
PanaFerd-AX | Free form | 3S,3′S | 2180 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| CYP2AE2 | Zebra fish; | Danio albolineatus | (50.2% of total Car) | De novo |
CYP2 | Genetics Heterologous expression | synthesis | [556][557][556,557] | [329][330][329,330] | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Paracoccus sp. strain N81106 (NBRC 101723) (Agrobacterium auranticum) (w/. mutation) |
N/A | Free form and glycoside | 3S,3′S | ~800 (63.2% of total Car) |
De novo synthesis |
[331] | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| (Actinopterygii: Cypriniformes) | Brevundimonas sp. M7 (w/. mutation) |
N/A | Free form ** | 3S,3′ | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| CYP2J2 | S ** | Anole Lizards; | Anolis favillarum | 130 | CYP2 CYP2 | De novo Synthesis ** |
[186] | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Genetics | [ | 313 | ] | Sphingomonas astaxanthinifaciens TDMA-17 |
N/A | Free form | 3S,3′S ** | 96.0 (34.3% of total Car) |
De novo Synthesis ** |
[182 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| CYP2J6 | ] | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| (Reptilia: Iguania, (Lepidosauria)) | Paracoccus haeundaensis KCCM 10460 (Co-culture w/. Lactic Acid Bacteria) |
N/A | Free form | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| CrtS (CYP5139Q1) | 3 | S, | Phaffia Yeast; | 3′S ** | 82.1 | De novo synthesis |
Xanthophyllomyces dendrorhous | CYP3 | Heterologous expression | [212][213][558][212,213,558] | [332] | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Paracoccus bogoriensis BOG6T (DSM16578, LMG2279) | N/A | Free form | 3S,3′S | 40 (10.8% of total Car) |
De novo synthesis |
[183] | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| (Fungi: Basidiomycetes) | Brevundimonas spp. | N/A | Free form ** | 3S,3′S ** | 2.8~36.5 | De novo | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| CYP384A1 | Spider mites; | Tetranychus kanzawai | Synthesis ** | CYP3 | Genetics | [375] | [186] | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Sphingomicrobium astaxanthinifaciens CC-AMO-30B |
N/A | Free form | 3S,3′S ** | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| CYP383A1 | 4.0 | (Arthropoda: Chelicerata) | CYP3 | De novo | Synthesis ** |
[185] | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Putative | (Closest homologue of CYP384A) | Brevundimonas sp. strain SD212 (NBRC 101024) |
N/A | Free form | 3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| CYP3A80 | Sira poison Frog; | S, | 3′S | N/A | Ranitomeya sirensis
The respective number was quoted from the reference(s), and it may vary depending on the collection location and season.†, the presence of binding forms to carotenoproteins would not be mentioned in this table; ‡, the identification method of the compounds remains uncertain; *, the biosynthetic pathways have not been fully characterized; **, based on the information on close species/genus; N/A; not available; N/D; not detected.*** Since astaxanthin is diversely found in the skin, feathers, and retinas of birds, only the characteristic reports are described. ?; based on the information on close taxa. For the details of distribution in avian species, see the other review [314]. 2.3. Biosynthesis and Metabolism of Astaxanthin2.3.1. Overview of Carotenoid Biosynthetic Pathways in Bacteria, Fungi, and Higher PlantsThe biosynthesis of AX is entirely based on β-carotene as the common precursor. Therefore, this document omits a detailed discussion of the metabolic pathway to β-carotene. Briefly, carotenoids belong to isoprenoids, which are the most diverse group of natural compounds. Isoprenoids are commonly biosynthesized from isopentenyl diphosphate (IPP). IPP is synthesized via the mevalonate pathway, which is present in almost all eukaryotes (the domain Eukarya) and archaea (the domain Archaea), as well as some actinobacteria (the phylum Actinomycetota). Alternatively, the MEP (2-C-methyl-D-erythritol 4-phosphate) pathway is used, which begins with pyruvate and glyceraldehyde 3-phosphate and proceeds through 1-deoxy-D-xylulose 5-phosphate (DXP) and MEP. This pathway is found in almost all bacteria and the chloroplasts of photosynthetic eukaryotes. IPP is isomerized to dimethylallyl diphosphate (DMAPP) through the action of IPP isomerase (Idi; IDI). Subsequently, DMAPP is converted to farnesyl diphosphate (FPP) and geranylgeranyl diphosphate (GGPP) through sequential condensation reactions with IPP. These reactions are catalyzed by FPP synthase and GGPP synthase, respectively [397]. Figure 810 illustrates the biosynthetic pathway of carotenoids from FPP in the leaves of higher plants, highlighting the enzyme-catalyzed reactions involved. Additionally, this figure presents the functions of carotenoid biosynthesis enzymes derived from bacteria and fungi [154][155]. According to past reports, the group of enzymes involved in the carotenoid synthesis pathway in green algae is composed of a set of genes that share homology with those of higher plants [398]. Figure 810. Biosynthetic pathway of carotenoids in the leaves of higher plants. Plant-type enzymes are shown with green letters, while bacterial enzymes and fungal enzymes that can catalyze in this pathway are written in pink and red, respectively.
2.3.2. Overview of Astaxanthin Biosynthetic Pathways in Bacteria, Algae and PlantsThe β-C3-hydroxylation and β-C4-ketoxylation reactions involved in AX biosynthesis are present in a wide taxonomic range of organisms. These reactions are mainly catalyzed by membrane-integral, diiron, nonheme oxygenase superfamily enzymes, and occasionally heme-dependent cytochrome P450-type monooxygenase enzymes. Detailed information regarding these enzymes is provided below. In Proteobacteria, such as the genera Paracoccus and Brevundimonas, the genes responsible for AX biosynthesis are well understood (refer to Section 2.2.1). AX is synthesized from β-carotene through the coordinated actions of CrtZ-type β-C3-hydroxylase (β-carotene 3,3′-hydroxylase; CrtZ) (EC: 1.14.15.24) and CrtW-type β-C4-ketolase (β-carotene 4,4′-oxygenase; CrtW) (EC: 1.14.99.63; including an incorrect description on the substrate specificity) with β-carotene as the initial substrate (see Figure 78 and Figure 911) [37][399][37,399]. However, due to variations in enzyme reactivity among different species, AX-producing bacteria often accumulate significant amounts of precursors, especially adonixanthin. Figure 911. Conversion routes from β-ring to 3-hydroxy-4-keto-β-ring in AX-biosynthesizing organisms. ASY, also called CrtS, from Xanthophyllomyces dendrorhous; CrtW and CrtZ, from bacteria; BKT and BHY, from green algae; CBFD and HBFD, from Adonis aestivalis.
2.3.3. How Can Hematococcus Algae Achieve Ultra-High Concentrations of Astaxanthin Biosynthesis?Haematococcus algae exhibit remarkable potential for accumulating up to 9.8% of AX in cell weight under specific culture conditions [328]. These AX compounds predominantly exist as mono- and diesters, gracefully stored within oil droplets housed in the resilient cyst cells, as discussed earlier (refer to Section 2.2.2). Notably, mature aplanospores (cysts) showcase the presence of AX primarily within intracytoplasmic oil droplets rather than plastid-derived chromoplasts [223][247][248][249][250][251] [223,247,248,249,250,251]. These specialized droplets serve a dual purpose, shielding the nucleus as antioxidants and acting as light filters, effectively reducing excitation pressure on photosynthetic subunits and mitigating the risk of photodamage [405].How is it possible for Haematococcus algae to achieve such a high concentration of AX compared to other organisms? One significant reason for this is their ability to accumulate the majority of AX as oil droplets in oil vacuoles within the cytoplasm rather than in plastids. These oil droplets, protein-lined structures typically synthesized from the endoplasmic reticulum, efficiently accumulate lipids (Figure 103), as observed in mammalian adipocytes and other organisms. Another crucial factor is the ester form of AX. Free AX, in the absence of a carrier protein, can aggregate at a certain soluble concentration, even in the presence of lipids as a solvent, potentially imposing physical stress on the cells. However, as fatty acid esters, the solubility of AX in fats, particularly triglycerides, is greatly enhanced. The biosynthesis and accumulation of AX and triglycerides within oil droplets occur in a coordinated manner, with de novo synthesis of free fatty acids and AX esterification taking place sequentially along this pathway (Figure 103) [406][407][408][409][410][406,407,408,409,410]. Consequently, Haematococcus algae have the capability to accumulate AX within oil droplets in the cytoplasm while the volume of the chloroplasts decreases.
 Figure 103. Pathways and localization of astaxanthin biosynthesis and esterification in Haematococcus algae. ACCase, acetyl CoA carboxylase; DGAT, diacylglycerol acyltransferase; FAS, fatty acid synthase; FA, fatty acid; SAD, stearoyl acyl carrier protein desaturase; TAG, triacylglycerol. Other enzyme abbreviations are listed in the main text. This figure was reproduced from ref. [410] with the permission of the publisher.
2.3.4. Metabolism of Astaxanthin in AnimalsOverviews of Metabolism of Astaxanthin in Animals; General RemarksIn animals, the accumulation of carotenoids, including astaxanthin (AX), is the result of complex food chains and metabolism. As depicted in Figure 114, carotenoids are biosynthesized in organisms at the beginning of the food chain. These organisms serve as prey for higher-level organisms, which selectively accumulate and metabolize carotenoids, including precursor forms and AX. Concurrently, AX can undergo metabolic processes, leading to the formation of different carotenoids or degradation. This section aims to present the most recent findings concerning the metabolism of AX in non-mammalian animals.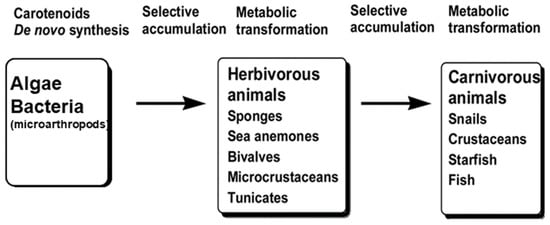 Figure 114.
Metabolic Conversion to Astaxanthin by Cytochrome P450In terrestrial animals, AX and its related “red” ketocarotenoids are commonly found in the retina and serve as ornamental coloration on the skin and plumage of reptiles and birds, as discussed in Section 2.2Section 2.2.5.5. The precise role of AX in these organisms remains hypothetical; however, it is clear that AX accumulation is derived from carotenoids present in their food through the food chain. In the case of fish, crustaceans, and flamingos, it has long been known that AX is converted from “yellow” carotenoids such as β-carotene and zeaxanthin, which can be precursors obtained from their diet. However, the specific enzymes involved in this conversion have remained unclear (Section 2.2.5). Recent studies have suggested an intriguing possibility that the red color characteristic of these organisms may also arise from the conversion of “yellow” carotenoids, such as β-carotene and zeaxanthin, by certain cytochrome P450 (CYP; P450) (refer to Table 3). The involvement of P450s in AX biosynthesis has been demonstrated through the analysis of ASY (CrtS) in Phaffia yeast, as discussed in SectionSection 2.3.1. 2.3.1. Notably, the P450s presumed to be involved in the metabolic conversion to AX in the animal kingdom are diverse and do not have direct orthologs to ASY (CrtS). In their study, Mundy et al. made a groundbreaking discovery by identifying a P450 gene (CYP2J19) that is potentially involved in the biosynthesis of “red” ketocarotenoids for the first time in the animal kingdom. They accomplished this through a comprehensive genetic analysis of the “yellowbeak” mutant of the zebra finch (Taeniopygia guttata) [552]. The yellowbeak mutant possesses a mutation in CYP2J19 within the gene cluster encoding CYP2, where the wild type exhibits CYP2J19A and CYP2J19B, whereas the yellowbeak mutant has CYP2J19yb. Consequently, CYP2J19A is exclusively expressed in the retina, while CYP2J19B is expressed in the beak and tarsus, with varying levels of expression in the retina. Conversely, CYP2J19yb expression is barely detectable in the beak of yellowbeak birds. These findings establish the essential role of CYP2J19 in ketocarotenoid biosynthesis in zebra finches [552]. Furthermore, Lopes et al., who belong to the same research group as mentioned above, reported the whole genome sequencing of red siskins (Spinus cucullata, exhibiting red body color), common canaries (Serinus canaria, exhibiting yellow body color), and “red factor” canaries (a crossbreed of red siskins and canaries) to investigate the genetic basis of red coloration in birds [553]. Their research identified two genomic regions crucial for red coloration in “red factor” canaries, one of which contains the gene encoding CYP2J19. Transcriptome analysis revealed a significant upregulation of CYP2J19 in the skin and liver of red factor canaries, further suggesting that CYP2J19 functions as a β-C4-ketolase, catalyzing the conversion of ketocarotenoids in birds [553]. It is plausible to assume that these P450 proteins acquired their functions through convergent evolution within each respective order/suborder [554]. While the metabolic pathways of these AX-generating P450s are expected to be similar to those of Phaffia yeast, information regarding the substrates involved in each P450 reaction and the absolute configuration of the resulting C3, C3′positions remains limited. The available data suggests that Acari, an organism capable of producing AX, exhibits an optical configuration of (3S,3′S), which is the opposite of the (3R,3’R) configuration observed in Phaffia yeast, as reported in an earlier publication. Therefore, it is anticipated that future studies will shed light on the reaction mechanisms, the substrate specificity of each P450 involved in AX conversion, the optical configuration of the produced AX, and the intermediates formed during the reaction. The CYP2J19 gene, generally present as a single copy in their genome, is widespread among avian lineages. This observation aligns with the notion of a conserved ancestral function in color vision, followed by subsequent co-selection for red epidermis coloration. Similar to several other CYP loci with conserved functions, CYP2J19 exhibits evidence of having undergone positive selection across bird species. Although there is no direct evidence indicating changes in selection pressure on CYP2J19 associated with co-selection for red pigment, it is possible that compensatory mutations related to selection on the adjacent gene CYP2J40 may contribute to this phenomenon [321]. In contrast, as discussed in Section 2.2.4, certain birds, such as penguins, kiwi, and some owls, lack ketocarotenoid-containing cone oil droplets in their retinas. In these avian species, the CYP2J19 gene has undergone pseudogenization [320]. Although penguins do contain AX in their body tissues, it is believed to be derived from their diet [395][396][395,396]. Interestingly, there have been reports indicating that some bird species may have lost and subsequently regained the function of CYP2J19 during the course of evolution, suggesting the potential advantageous significance of reddish body coloration and the acquisition of ketocarotenoids for birds [555]. The zebra finch, for which the function of the CYP2J19 gene was first indicated, also has two copies of the CYP2J19 gene that have probably been duplicated during evolution [552]. Among tetrapods, turtles are the only group that possesses red oil droplets in their retinas, and several turtle species exhibit red carotenoid coloration. Twyman et al. conducted a study on the evolution of CYP2J19, a gene associated with color vision and red pigmentation in reptiles, utilizing genomics and gene expression analysis. They discovered that turtles, but not crocodilians and lepidosaurs (including lizards, snakes, and tuatara), possess orthologs of CYP2J19, which originated from a gene duplication event prior to the divergence of turtles and archosaurs. CYP2J19 is strongly and specifically expressed in the retina and red outer skin of turtles, which include ketocarotenoids. The researchers propose that CYP2J19 initially played a role in the color vision of archosaurs, and they conclude that red ketocarotenoid-based coloration independently evolved in birds and turtles through genetic regulation changes involving CYP2J19. In other words, these intriguing findings suggest that red ketocarotenoids might have contributed to color vision and ornamental coloration in dinosaurs and pterosaurs [554]! Therefore, the presence of CYP2J19 genes plays a crucial role in the ketolation of carotenoids in avian and reptilian species. However, reptiles, particularly lepidosaurs, likely lack CYP2J19 [554]. Despite this, several reptile species exhibit ketocarotenoids, such as AX, in their integumentary coloration (refer to Section 2.2.4. for more details). Subsequently, a genetic approach identified genes involved in the conversion of carotenoids to ketocarotenoids in spider mites and a species of zebrafish [375][556][375,556]. Interestingly, these P450-type C4-ketolase enzymes in spider mites and zebrafish are not encoded by CYP2J19 but are shown to be associated with separate P450 enzyme genes. For instance, in spider mites (Tetranychus kanzawai), the P450 encoded by the CYP384A1 gene significantly contributes to red body coloration through ketocarotenoids [375]. Similarly, in the zebrafish species (Danio albolineatus), the CYP2AE2 gene is involved in the accumulation of red pigment in the skin erythrophores [556][557][556,557]. In other organisms, genetic studies and homology with known carotenoid oxygenases indicate the involvement of various P450 enzymes in the β-C4 oxygenation of carotenoids (Table 3). For example, Anolis favillarum, a reptile belonging to the lepidosauria (lizards), exhibits white, yellow, and orange skin. The orange skin coloration is attributed to pteridines and ketocarotenoids. As mentioned earlier, reptilian lepidosauria have lost CYP2J19 during evolution [N/D; not determined.
2.4. Chemical Synthesis and Analysis of Astaxanthin2.4.1. Chemical Synthesis of AstaxanthinIn this document, while the main focus is on AX derived from biological sources, a brief overview of chemically synthesized AX will also be provided. In 1967, Surmatis et al. were the first to synthesize AX in its dimethyl ester form [564]. I In the same year, Leftwick and Weedon successfully synthesized the free form of AX from canthaxanthin [565]. In the early 1980s, the group at Hoffman-LaRoche achieved the total synthesis of astaxanthin through the Wittig reaction of C15-end-group phosphonium salts at both ends of the central C10-dialdehyde (Figure 125). Using this method, they synthesized (3S,3′S), meso, and (3R,3′R) optical isomers of AX, along with several geometric isomers [566]. This synthetic method was employed for industrial synthesis, and synthetic AX is still commercially available as a coloring agent in aquaculture for fish such as salmon, trout, and sea bream. It is important to note that commercial products (Carophyll Pink or Lucantin Pink) are coated with gelatin and starch, making them water-dispersible. Figure 125. Industrial synthesis route for astaxanthin.
2.4.2. Quantitative and Qualitative Analysis of AstaxanthinPractical analysis of AX is primarily conducted using UV/VIS absorbance spectrophotometry and/or HPLC methods. The absorbance spectrophotometric method is simple and cost-effective when analyzing samples that predominantly contain AX as the carotenoid. The molecular absorption coefficient 𝐸1%𝑐𝑚 = 2100 (in acetone, hexane, and EtOH) of AX is often utilized [4][68][4,68]. HPLC systems are commonly employed for the quantification and identification of AX in carotenoid mixtures. Reversed-phase systems using ODS or C-30 columns are frequently used for quantitative and LC-MS analysis [64][68][264][568][64,68,264,568]. However, normal-phase systems with silica gel columns or CN columns can also be employed. The normal-phase system offers superior separation efficiency for analogues with similar polarity and geometrical isomers [56][181][187][196][401][56,181,187,196,401]. Chiral columns such as Sumichiral OA-2000 can be used for the analysis of optical isomers [53]. Commercially available, highly purified AX can be used as a standard; however, if the origin of the standard is synthetic, it typically consists of a mixture of the three optical isomers, unless otherwise specified. Standard AX esters from Haematococcus algae can be purchased from the USP with validated concentrations [68]. Depending on the analytical environment, however, it has been reported that the quantitative value may not be consistent due to the influence of light and ambiguous other factors [569]. This is considered to be mainly due to the fact that 13- and 15-cis geometrical isomers, which are bent around the center of the molecule, are easily converted to all-trans isomers by light [56][147][56,148]. Therefore, it is considered necessary to standardize the conditions for quantification, such as performing the analysis under light-shielded conditions. For another reason, geometric isomers with two or more of the molecule’s double bonds arranged in the cis configuration (e.g., di-cis isomers) have not been included in the quantitative values by the current USP method. Since these “multi”-cis molecules are also affected by light, it is possible that the quantitative value is estimated to be low by the current method. Therefore, the development of an improved analytical method should accelerate in the near future. When AX presents as its fatty acid esters (See Section 2.1.2), it must be hydrolyzed by cholesterol esterase or lipase to lead to the free form for identification and/or quantification. For example, in the case of quantitative analysis of AX in Haematococcus algae, the analytical method was validated using cholesterol esterase from Pseudomonas sp. (EC 3.1.1.13, available from Merck (Sigma-Aldrich) or FUJIFILM Wako Chemicals) [68]. In some instances, an inexpensive lipase powder from Candida cylindracea (EC 3.1.1.3, Lipase OF, Meito Sangyo Co., Ltd., Japan) [265] was used for qualitative analysis, showing comparable activity to cholesterol esterase. Roche’s group employed alkali saponification under completely anoxic conditions to saponify the fatty acid esters of AX, leading to free AX; however, the experimental setup and operation were highly complex [570]. Preparative liquid separation is often required for the purification of post-saponification treatments and biological samples. In such cases, the use of an internal standard (IS) with similar physical properties is crucial. Preferred IS options include Ehinenone, Ethyl 8′-apo-β-caroten-8′-oate, and trans-β-apo-8′-carotenal [68][568][571][572][68,568,571,572]. HPLC allows for the separation of all-trans forms of AX and geometrical isomers such as 9-cis, 13-cis, and 15-cis AX. These isomers exhibit different UV-VIS spectra and can be easily identified by comparison with reference values if the HPLC system includes a PDA detector [4]. More recently, LC-MS with APCI and ESI ionization, along with UV-VIS detection, has been used for the detection of AX during separation using HPLC systems. AX exhibits a prominent protonated molecule (MH+) with m/z 597 in APCI and ESI-MS. The detection sensitivity of carotenoids by LC-APCI-MS and LC-ESI-MS has significantly improved to the sub-ng order. LC/MS (or MS/MS) is also a powerful tool for the analysis of complex mixtures such as AX fatty acid esters and AX glycosides [64][265][64,265]. While HPLC analysis is accurate and reliable, it involves complex procedures and requires skill for analysis. In recent years, the use of resonance Raman spectroscopy has been considered as a potential solution for more noninvasive, on-site analysis. The resonance Raman spectra of AX, when excited by visible lasers, exhibit dominant bands at approximately 1008 cm−1, 1158 cm−1, and 1520 cm−1 [573]. Raman spectra measured on samples of salmon fillets at fish markets have demonstrated the detection of AX in salmon skeletal muscle [129]. Portable Raman spectrometers have become available, allowing for on-site studies, and it has been reported that the signal intensity of the AX-specific Raman band at 1518 cm−1 (C=C stretching frequency) increases in an AX concentration-dependent manner. This signal can be distinguished from fish proteins and lipids, enabling the determination of AX levels in different parts of salmon fillets and different species of salmon. These findings indicate the effectiveness of Raman spectrometry for on-site AX quantification [574]. Polynomial approximation or multivariate curve resolution-alternating least squares (MCR-ALS) using the Raman spectrum has also been used for the quantification of salmon, Phaffia yeast, and Haematococcus algae [251][575][576][577][251,575,576,577]. Raman microscopy has shown great potential for diverse types of analysis. AX, due to its strong lipophilic properties, co-localizes with lipid fractions in cells and exhibits a characteristically strong Raman spectrum. Therefore, it has been investigated as a non-toxic, non-destructive Raman probe for organelles [578][579][578,579]. Pioneering studies have used resonance Raman microscopy to determine the localization of topically applied AX in skin tissue [580]. It appears that the methods mentioned in these reports can also detect analogues (such as canthaxanthin and adonirubin) and degradation products [581], although these are not major concerns when the purity of AX is relatively high and the variation in the profile of these impurities is minimal. In conclusion, the selection of an appropriate analytical method is crucial and depends on the specific requirements of the analysis.2.5. Relationship between Human Culture and Astaxanthin2.5.1. Historical Exposure of Human Societies to Astaxanthin Sources in NatureHuman societies have interacted with nature for thousands of generations and taken advantage of substances with medical and health benefits. Carotenoids are the most abundant family of pigments in nature. Aquatic resources create numerous opportunities for humans to experience various applications of natural pigments, including AX [582]. AXs namesakes, the crayfish and lobster, hold an important place in many culinary and cultural traditions, including those of Sweden and America. Astacus astacus is a European crayfish species native to Scandinavia, that become popular among Swedish nobles in the Middle Ages. By the 16th century, festive crayfish parties became a Swedish tradition, primarily featuring freshwater crayfish (flodkräfta) or marine lobster (Nephrops norvegicus) in the case of the Swedish west coast. Both crayfish and lobster turn bright red after boiling when AX is released from a protein complex in the crustaceans’ shell. The celebration was coined “kräftskiva” in the 1930s and is still celebrated to-day in the month of August [583][584][583,584]. The luxury and popularity of AX-rich crustacean meals were often depicted in the still-life paintings of the Dutch Golden Age. The bright oranges and reds of cooked crustaceans were centerpieces in paintings depicting light breakfasts called “ontbijtjes” and lavish banquets featuring objects of luxury, called “banketje” or “pronkstilleven” [584]. In the United States, the red swamp crayfish (Procambarus clarkii) is popular in the state of Louisiana, where crayfish are also known as crawfish, mudbugs, and crawdads. The crawfish is a central figure in the culture of Louisiana’s Native American Houma nation. He was once part of the Chakchiuma group, distantly related to both the Choctaw and Chickasaw. “Shâkti Humma”, or “red crawfish”, was a giant crawfish who created the world and formed living creatures out of mud. Houma warriors depicted the red crayfish as their war emblem because this tiny creature is known to brandish its pincers, never backing down, even in the face of larger enemies. The red crawfish was an integral symbol of resilience and strength, and even the word “Hou-ma” means “red” [585][586][587][585,586,587]. When the Acadians from the Canadian maritime provinces settled in the bayous of Louisiana in the 17th century, they may have observed the Houma harvesting crawfish. The Acadians, now called Cajuns, may have adapted their Canadian lobster recipes for making crawfish boils, which today form the backbone of Louisiana cuisine. In the 1980s, the crawfish officially became Louisiana’s state crustacean, further reinforcing the cultural significance of AX in this region [588]. Although crustaceans are a celebrated source of AX, the most abundant source of AX in the human diet is wild salmon. Salmon have been an important food source for coastal cultures across the Northern Hemisphere since prehistoric times and are widely featured in legends and depicted in art. The oldest known image of salmon may be a relief carving of a salmon in a cave in France from the Gravettian era (25,000–20,000 BCE) [589][590][589,590]. Salmon is revered in many parts of the world. In Native American art, salmon are a symbol of perseverance, ancestral knowledge, and spiritual journey. The First Salmon Ceremonies practiced across the American Northwest celebrate the arrival of spawning salmon each spring [591]. In Celtic mythology, Fionn mac Cumhaill, the legendary giant credited with constructing Northern Ireland’s famous Giant’s Causeway, gained knowledge of the world by tasting the Salmon of Wisdom [592]. One of the tales from the 6th century featuring King Arthur recounts his quest, during which he rode on the back of a salmon to Gloucester and freed a captive deity [593]. In Icelandic Norse mythology from the 13th century, the trickster deity Loki transformed into a salmon to escape his brother Thor. However, Thor caught Loki at the base of his tail, forming the iconic grip point for fishermen [593]. In the city of Murakami in Niigata Prefecture, Japan, salted and wind-dried salmon have been prepared using traditional methods since the Heian period (794 to 1185). The history of salmon consumption in the region can be traced back even further, with salmon bones found in archaeological remains from the Jomon era (pre-B.C.). Salmon has long been recognized as one of the most important foods in the region. During the Edo period (1603–1867), when Murakami served as a feudal domain under the Tokugawa shogunate, salmon held significant economic value, serving as a currency for paying taxes, presenting gifts to the Imperial Court and Tokugawa shogunate, and being distributed as salaries to government officials. In the late Edo period, when salmon catches in the Miomotegawa River flowing through the city declined, a samurai named Heiji Aotobu proposed a salmon propagation project to the domain government. River construction and resource conservation efforts were undertaken to create an environment conducive to salmon spawning, resulting in the recovery of salmon populations. This work was groundbreaking in terms of sustainable fishing practices. Presently, Murakami salmon continues to be presented as gifts and is featured in special meals during New Year celebrations [594]. Murakami’s salted salmon has also been used in the Daijosai, the first Niiname-sai (a Japanese harvest ritual) of a Japanese emperor following their enthronement. This once-in-a-generation festival involves the emperor serving the newly harvested grains to the gods of heaven and earth, “Amaterasu and Tenjin Chigi”, as well as partaking in them. The festival site, known as “Yuki” in the east and “Suki” in the west, receives harvested grains from all over Japan to be offered to the gods. Murakami’s salted salmon is among the products from Niigata Prefecture dedicated to Yuki [595]. The Ainu people, who have inhabited northern parts of Japan since ancient times, also consider salmon to be a vital dietary source. The Ainu refer to salmon as “kamuichep” (divine fish) or “shipe” (the true food). The Ainu hold a “new salmon prayer” called “Asiri Chep-nomi” when the salmon return, seeking a bountiful catch. They have various rituals and traditions associated with salmon, which skillfully reflect the salmon’s ecology and the wisdom of their ancestors. For example, the first salmon to return to the river is reserved for the fox gods, who protect the water source, and must not be caught. The subsequent salmon are designated for other gods and then shared among humans, who rely on the salmon for sustenance [596][597][596,597]. Moreover, salmon holds deep cultural and culinary significance not only for the Ainu but also for the indigenous peoples of the North Pacific [596]. While animal sources continue to inspire and sustain AX consumption, the increasing demand for AX as a nutritional supplement must not become a burden on aquatic ecosystems. As exemplified by samurai Heiji Aotobu, environmental conservation and stewardship must guide towards a sustainable future. That is why we turn to Haematococcus algae as a sustainable source of natural AX.2.5.2. Human Culture Shift towards Sustainability: Haematococcus algae as a Promising Source of Natural AstaxanthinAs discussed, historical evidence suggests that indigenous or traditional knowledge, such as knowledge about food and medicine, was developed through human interaction with the environment and nature. This knowledge has been passed down through generations as an integral part of human culture. In addition to aquatic animals such as shrimp and salmon, the consumption of freshwater aquatic plants has ancient origins due to their abundance, unique sensory properties, and nutritional and health benefits [598]. Microalgae and seaweed have been used as food since medieval times and have established markets in Asia, with a growing market in Europe [599]. Archaeological findings from Monte Verde, Chile, indicate that the use of marine algae as food dates back around 14,000 years [600]. Not only were aquatic plants consumed as food, but plant-based medicinal extracts, including algae, herbs, and fungi, were also prevalent. This knowledge forms the foundation for many contemporary health-related discoveries. In fact, over the past century, eating and feeding practices have evolved from social and cultural customs to explicit healthcare and medical practices [601]. Modern nutritional science has shed light on the nutritional and therapeutic effects of various components found in aquatic plants and their role in biological processes. Furthermore, it is essential not to overlook the connection between the health of human populations and the Earth’s natural food systems and biodiversity, as well as the role of microalgae in ecosystem preservation. In the late 18th century, Alexander von Humboldt, an influential German scientist, was one of the first to note the negative impact of human-induced environmental alterations (although the terms “ecology” and “ecosystems” did not exist at that time) and extensive land use on human well-being [602]. Overexploitation (harvesting species from the wild at rates that exceed their reproductive capacity) and climate change have been identified as major drivers of biodiversity decline on Earth [603]. In one of the latest reports by the European Commission (EC), developed through collaboration between 50 experts from 25 EU Member States, a set of recommendations has been formulated to rethink the relationship between humans and nature, highlighting the role of human culture in achieving the Sustainable Development Goals (SDGs) [604]. Microalgae, in this regard, are being considered as a promising candidate that can directly or indirectly contribute to the SDGs [605], while also having historical roots in human culture. As carbon-hungry and nutrient-rich “sustainable biofactories”, microalgae offer potential solutions for global food security and mitigating environmental issues [606][607][606,607]. The historical roots of microalgae and its derivatives in human culture facilitate the reintroduction of algal biomass and algal bioactive functional ingredients, including AX, into global food and nutrition systems. Over the past few decades, studies on microalgae and their bioactive compounds, such as AX, have continuously revealed their role in promoting the circular economy and their positive effects on the health and well-being of the Earth and its flora and fauna. Although Haematococcus alga was discovered in the 18th century [222], its significance as a promising sustainable and abundant natural source of AX has garnered increasing attention in recent years. It is important to note that overexploitation of any flora and fauna, including algae and other carotenoids’ sources in nature (fruits and vegetables), may have an irreversible impact on the environment. In this case, well-designed and properly scaled commercial/artificial mass production of carotenoids seems to be more sustainable. In fact, sustainability potential includes the source from which AX and other carotenoids are obtained. Microalgae is considered a more sustainable source than other plant sources of carotenoids. For instance, the marigold flower is currently the predominant natural source of lutein. However, it has a lower growth rate than microalgae and requires arable land, and its harvest and extraction are only seasonal. Such limitations apply to other carotenoids from fruits and vegetables, such as zeaxanthin. There are some concerns about the sustainability of agricultural products as compared to algaculture, including the use of water, land, pesticides, and fertilizers. Microalgal cultivation has added environmental benefits over plants with higher carbon sequestration, a reduced water footprint, and no pesticide use. Both lutein and AX from microalgae can be considered sustainable active compounds, with even potentially complementary health benefits, especially for vision and eyes [608]. Currently, there is no commercial production of lutein from microalgae [609][610][609,610]. Moreover, although this review is focused on AX, it is noteworthy that this phytonutrient is only part of the algal meal. The whole microalgae biomass is consumable by humans and contains several high-value nutrients and bioactive molecules with health-promoting properties, including protein, essential fatty acids, antioxidant compounds, etc. For this reason, there is normally no or negligible amount of waste or residue produced from microalgae harvest, unlike other carotenoids extracted from agricultural products. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||