Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Jessie Wu and Version 1 by José A Sanchez Alcazar.
The term neurodegeneration with brain iron accumulation (NBIA) brings together a broad set of progressive and disabling neurological genetic disorders in which iron is deposited preferentially in certain areas of the brain. Among NBIA disorders, the most frequent subtype is pantothenate kinase-associated neurodegeneration (PKAN) caused by pathologic variants in the PANK2 gene codifying the enzyme pantothenate kinase 2 (PANK2).
- neurodegeneration with brain iron accumulation (NBIA)
- pantothenate kinase-associated neurodegeneration (PKAN)
- pantothenate kinase 2 (PANK2)
- pantothenate
1. Introduction
Neurodegeneration with brain iron accumulation (NBIA) represents a group of rare genetic neurodegenerative diseases that clinically manifest the presence of severe dystonia, rigidity, dysarthria, loss of ambulation, parkinsonism, choreathetotic movements, retinal degeneration or optic nerve atrophy, neuropsychiatric disorders and can lead to premature mortality [1]. The most frequent pathological findings are iron deposits in the basal ganglia and adjacent areas, and generalized axonal dilations (called spheroid bodies) in the central nervous system (CNS), representing degenerated neurons [2]. At present, more than 15 genes are associated with NBIA disorders [3]. However, the responsive genes of nearly 20% of the patients with clinical suspicion of NBIA are unknown.
Despite the intense efforts in research on these diseases and the proposals of new therapeutic approaches, there are still no effective treatments to halt the progression of neurodegeneration in NBIAs. Therefore, new therapeutic strategies are necessary.
Pathological variants in the pantothenate kinase 2 (PANK2) gene, which encodes for an essential enzyme involved in the coenzyme A (CoA) biosynthesis pathway, are one of the most prevalent NBIA subtypes; it represents nearly 50% of cases [4]. Pantothenate kinase-associated neurodegeneration (PKAN) includes a continuous phenotypic spectrum with two major clinical forms: classic PKAN and atypical PKAN. Classic PKAN has an early onset in childhood (usually in the first decade of life) and a rapid neurodegenerative progression. On the other hand, atypical PKAN is characterized by a later onset (commonly in the second or third decade of life), and a slower course of the disease [5,6][5][6]. Despite this clinical classification, there are patients with early disease debut but insidious progression or late start with fast progression.
The PANK gene family comprises PANK1a, PANK1b, PANK2, PANK 3 and PKAN4 genes, but only pathological variants of PANK2 cause PKAN. PANK1, PANK2, and PANK3 isoenzymes are active as dimeric complexes with different localizations in the cell. PANK2 is the only isoform to be expressed in mitochondria in humans and primates [7], whereas PANK1 and PANK3 are commonly localized in the cytosol and nucleus [8]. On the other hand, PANK4 is a pseudo-pantothenate kinase that lacks kinase activity; however, it shows phosphatase activity catalyzing the dephosphorylation of phosphopantothenate, 4′-phosphopantetheine and its derivatives [9,10][9][10].
The PANK2 enzyme catalyzes the key regulatory reaction in CoA biosynthesis in which pantothenate is converted into 4′-phosphopantothenate using ATP. The main mechanism for controlling PANK2 activity is through allosteric inhibition by acetyl-CoA and CoA thioesters [11].
2. Therapeutic Strategies
At present no efficient therapy is available for PKAN. Thus, current treatments are aimed at controlling patient symptoms [1]. Although clinical trials with several compounds are in progress, PKAN treatments primarily aim to control the main disease symptoms: spasticity, seizures, dystonia, or psychiatric disorders [124][12]. Nevertheless, several promising therapeutic approaches are currently in progress [12,91][13][14]. These treatments can be summarized in four categories: (1) iron chelation to eliminate iron accumulation in the brain; (2) metabolite supplementation to correct metabolic deficits in the CoA pathway; (3) PANK isoforms activation to restore CoA biosynthesis; and (4) gene therapy by introducing the wild-type PANK2 gene. However, some of these therapies have not been successful, whereas others are under evaluation. For a detailed updated of current PKAN treatment approaches see [12,91,124,125][12][13][14][15].
It is noteworthy that despite the importance of autophagy in neuronal homeostasis and pathological processes such as neurodegeneration [126][16], there are few studies addressing autophagy modulation in PKAN disease models. Recently, Huang et al. have shown that fumble (fbl), the human PANK2 homolog in Drosophila, interacts genetically with PINK1 (PTEN-induced putative protein kinase 1), a key protein involved in the selective autophagy of mitochondria (mitophagy) [127][17]. In addition, mitochondrial fumble overexpression rescued PINK1 loss-of-function defects such as mitochondrial dysfunction. Interestingly, vitamin B5 derivatives restored CoA/acetyl-CoA levels and mitochondrial function, reversing the PINK1 deficiency phenotype [127][17].
2.1. Strategy for Finding Alternative Treatments for Pantothenate KANinase-Associated Neurodegeneration Using Patient-Derived Cellular Models
A key finding to support the utility of cellular models in PKAN research was that the supplementation with pantothenate, the substrate for the PANK2 enzyme, was able to increase PANK2 expression levels in patient-derived fibroblasts carrying pathologic variants with residual enzyme levels [19][18]. Moreover, the pantothenate-mediated up regulation of PANK2 levels was accompanied by the correction of all pathological alterations associated with PKAN such as iron/lipofuscin overload, increased lipid peroxidation and impaired mitochondrial bioenergetics. Furthermore, the positive effect of pantothenate was confirmed in iNs generated by direct reprogramming of PKAN fibroblasts [19][18]. These observations suggest that cell models may be a useful tool to identify patients with PANK2 mutations that respond in vitro to pantothenate supplementation. More importantly, these observations support the possibility of their treatment with high doses of pantothenate. In addition, these results suggest that personalized screening strategies in PKAN may facilitate the detection of more pharmacological chaperones (PCs) capable of increasing and stabilizing the expression levels and activity of the mutant PANK2 enzyme in specific mutations.
Many mutations in human diseases provoke the destabilization of the mutant proteins. Curiously, compounds that work as PC can rescue the activity of unstable proteins [128,129,130][19][20][21]. However, individual patients will be only suitable for therapy with PC depending on their specific genotype [131][22]. Supporting this assumption, it has been shown that several PANK2 pathological variants, but not all, can be rescued by pantothenate [19][18]. Therefore, a strategy for selecting more positive PCs in PKAN cellular models can lead to the identification of potential therapeutic alternatives in patients harboring specific mutations. Following this approach, several rare diseases can be already treated with PCs [132][23]: For Gaucher disease, Diltiazem, an antihypertensive drug [133][24]; for cystic fibrosis, Doxorubicin, an anti-cancer anthracycline, for cystic fibrosis [134][25]; for Pompe disease, Acetylcysteine, a mucolytic agent [135][26]; for Fabry and Gaucher disease, Ambroxol, another mucolytic agent [136][27]; for hyperinsulinemic hypoglycemia, Carbamazepine and dibenzazepine, [137][28]; for GM2 gangliosidosis, Pyrimethamine, an anti-parasitic drug [138][29]; and for Pendred syndrome, Salicylate, a well-known anti-inflammatory agent [139][30]. For PKAN disease, an allosteric brain-permeable PANK activator (PZ-2891) has been found [84][31]. Interestingly, a knockout mouse model of brain CoA deficiency under PZ-2891 therapy showed weight gain, improved locomotor activity and extended life span [84][31]. The aim of this therapeutic approach is to compensate for the loss of PANK2 by the activating of the other PANK isoforms [84][31].
2.2. Precision Medicine in Pantothenate PKAKinase-Associated Neurodegeneration
Precision medicine is an emerging approach that considers the adaptation of clinical management to the genetic characteristics of each patient. Clinical precision medicine for the management of genetic neurodegenerative disorders seems a more rational strategy in contrast to the traditional “one drug fits all patients” approach [140][32]. In fact, genetic neurodegenerative diseases can present heterogeneous clinical characteristics even in patients carrying the same disease or pathological variant. Furthermore, as several metabolic or signaling pathways can be secondarily affected it is highly unlikely that patients can benefit from a single drug. Genetic neurological diseases are promising models for precision medicine due to the increasing knowledge of the genetic basis of the disease and clinical classification, the increased number of biomarkers, and the existence of possible disease-modifying treatments [141][33].
In this context, precision medicine strategies using patient-derived fibroblasts and iNs could help optimize therapeutic approaches in PKAN.
Strategies based on precision medicine are currently applied in different health disciplines such as cardiology, nutrition, and oncology, as well as in rare diseases [142,143][34][35]. In neurodegenerative diseases, the first approaches based on precision medicine have been more relevant in Alzheimer’s disease (AD). Thus, anti-amyloid-β monoclonal antibody therapy is now being tested in patients with mutations known to cause AD with the aim of preventing neurodegeneration in patients with similar genetic alterations (ClinicalTrials.gov number NCT01760005, accessed on 5 May 2023). In addition, APOE (apolipoprotein E) variants can identify individuals at higher risk for AD [144][36], making them interesting biomarkers for earlier diagnosis, and the implementation of treatment and/or prevention strategies. Today, Parkinson's Disease (PD) is treated as one clinical entity, but many researchers emphasise that PD encompasses different sub-groups that can benefit from the approaches of precision medicine [145][37]. However, the complex nature of PD and AD, together with clinical phenotypic heterogeneity, present significant challenges to successfully implementing personalized medicine in these diseases.
The main phases of a personalized medicine approach applied to PKAN are illustrated in Figure 1. First, a skin biopsy is performed to generate fibroblast cultures. Subsequently, fibroblasts are characterized by examining the main alterations of PKAN disease such as iron/lipofuscin accumulation, lipid peroxidation, senescent morphology, and mutant protein expression levels. In addition to verifying PANK2 function, the expression levels of downstream proteins such as mtACP are also evaluated. Next, pharmacological screening is carried out to identify the compounds capable of correcting the alterations detected. In parallel, induced neurons are generated by indirect or direct reprogramming, verifying that they express the neuronal markers. Finally, the positive compounds identified in the fibroblast screening are evaluated in the induced neurons.
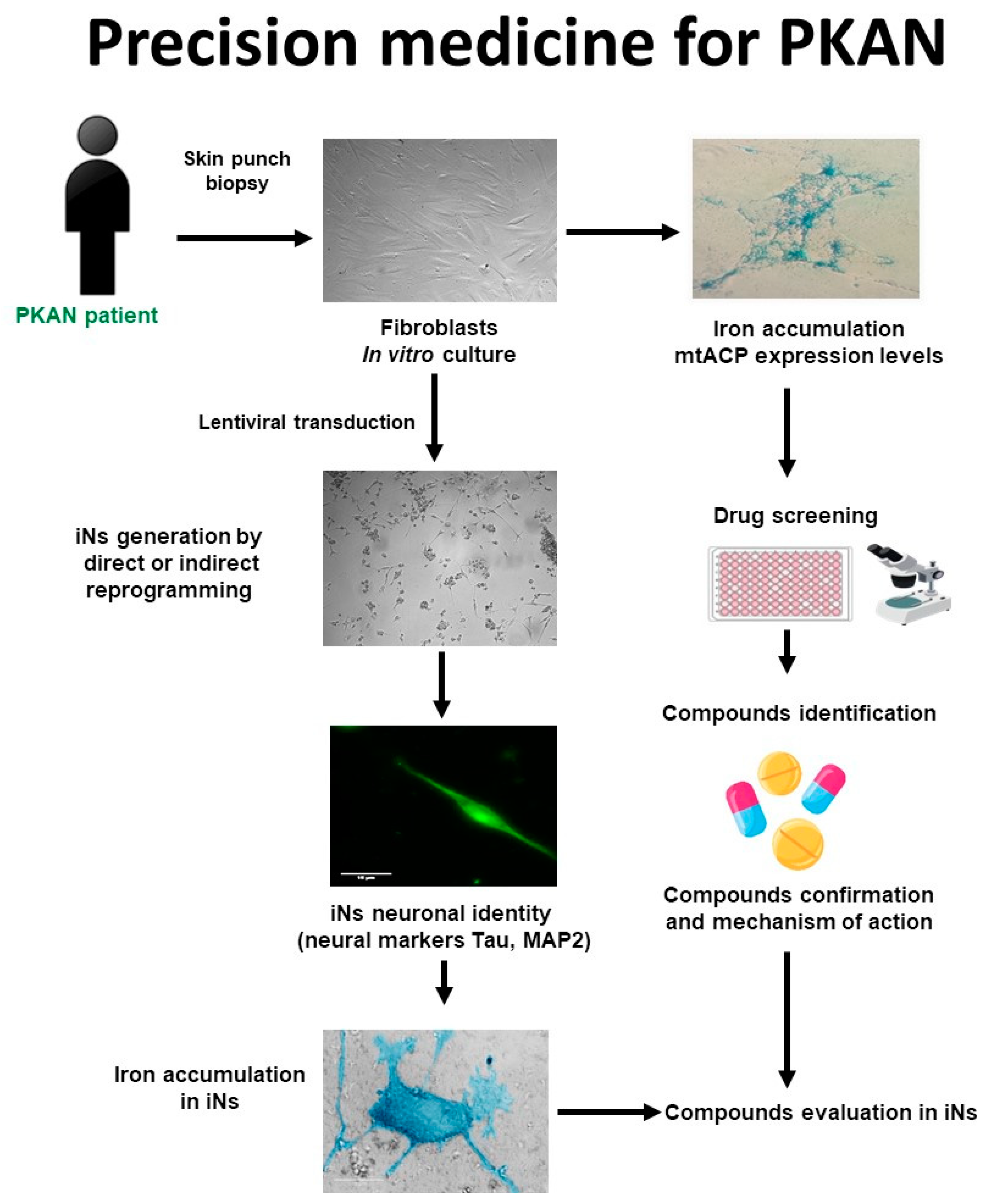
Figure 1. Cell-based disease modeling and drug screening approach in PKAN. PKAN patient-derived cellular models, fibroblasts, and iNs, can be useful tools for mimicking pathophysiological alterations of the disease and screening potential therapies. iNs = induced neurons; mtACP= mitochondrial acyl carrier protein.
Using this strategy, 7 positive commercial supplements (pantothenate, pantethine, vitamin E, omega 3, α-Lipoic acid, L-carnitine, and thiamine) have been recently identified [122,123][38][39]. All of them were able to eliminate iron/lipofuscin accumulation, increase PANK2 and mtACP protein levels, and correct the altered phenotype in responsive mutant cells.
The rationale of pantothenate supplementation assumes that mutant enzymes may function better with higher substrate concentrations. The ability of high-dose pantothenate supplementation to improve the activity of a functionally deficient PANK enzyme is supported by in vitro studies where the affinity of the enzyme for pantothenate can be low but the reaction is still functional [146][40]. These observations are interesting because they indicate that pantothenate supplementation at high doses may be clinically useful for patients carrying pathological variants with residual PANK2 expression levels and/or activity. However, this therapeutic strategy is not effective in patients carrying frameshift mutations causing termination codons in both alleles that encode the expression of an incomplete/truncated protein. For this reason, in vitro evaluation of the effect of pantothenate supplementation on patient-derived cells may provide valuable information on the response of specific pathological variant subgroups. Furthermore, it is necessary to check whether pantothenate treatment can reach the proper concentration to achieve the desired functional effects in the human brain in vivo. A strategy to solve this difficulty would be to perform combined treatments with pantothenate and other pantothenate derivatives such as pantethine with the aim of increasing pantothenate concentrations in the blood and in the brain.
Pantethine is a physiological compound synthesized from pantothenic acid and cysteamine, participating as a metabolic intermediate in the biosynthesis of CoA. Pantethine treatment can increase pantothenate levels in the blood because it is highly unstable, and it is rapidly transformed into pantothenate and cysteamine [147,148][41][42]. Pantethine supplementation has been shown to rescue PKAN phenotypes in several biological models such as bacteria [149][43], Drosophila [83][44], zebrafish [87][45] and mice [79][46]. The therapeutic potentiality of pantethine in PKAN has been mainly evaluated in animal models, although the compound has been used as a lipid-lowering agent in clinical studies [150][47]. Recently, the safety and efficacy of pantethine (60 mg/day during 6 months) in fifteen children with PKAN have been evaluated [151][48]. The conclusions of this restudy earch were that pantethine supplementation did not alter serum CoA levels or improve clinical symptoms. The poor therapeutic efficacy of pantethine in PKAN patients in this study research may be due to (1) the low number of patients under treatment; (2) the treatment duration was short; (3) a low dose concentration or low bioavailability of pantethine. However, as pantethine supplementation can increase blood pantothenate concentrations, the combination of both pantothenate and pantethine can be more efficient in specific patients.
Signs of oxidative and increased ROS production after iron exposure have been previously reported in PKAN cellular models [72][49]. Consistent with these findings, Alvarez-Cordoba et al., found increased content of carbonylated proteins and mitochondrial lipid peroxidation in PKAN fibroblasts [19][18]. Lipid peroxidation is generally described as a chain reaction caused by the oxidative damage of polyunsaturated fatty acids (PUFA) resulting in the generation of lipid peroxyl radicals, hydroperoxides and aldehyde derivatives [31][50]. Three stages are described during the process of lipid peroxidation: initiation, propagation, and termination [152][51]. The chemical reactions associated with each of these steps can be found elsewhere [153][52]. Peroxidation of lipids can disturb the assembly of the membrane, causing alterations in fluidity, permeability and ion transport [154][53]. Furthermore, many breakdown metabolites, such as malondialdehyde (MDA) and 4-hydroxynonenal (4-HNE) are generated in this process [155][54]. MDA and 4-HNE protein and DNA adducts modify multiple cellular processes and participate in secondary crosslinking reactions which may worsen the pathophysiology of the disease. In addition, lipid aldehydes may affect protein kinases and phosphatase activities leading to the abnormal activity of various transcription factors involved in cellular homeostasis [156][55].
Lipid peroxidation in organelles with high iron content, such as mitochondria, and alteration in membrane-dependent cellular processes such as vesicle trafficking and/or autophagy/mitophagy, can cause iron accumulation in lipofuscin granules, which in turn increases lipid peroxidation of membranes [156][55]. This vicious cycle of events that augment each other may aggravate and precipitate the progression of neurodegenerative diseases such as PKAN. Membrane antioxidants, such as vitamin E, can block this vicious cycle in neurodegenerative diseases by stopping lipid peroxidation propagation [157][56].
In addition, vitamin E is a necessary nutrient for neural development and neurological function [158][57]. This fact, together with much evidence demonstrating that neurodegenerative diseases are associated with oxidative stress and lipid peroxidation, leads to the hypothesis that the progression of neurodegeneration may be mitigated by membrane antioxidants such as vitamin E [159][58]. Several works in human and animal models of vitamin E deficiency assessed its participation in protecting the brain, and more specifically the cerebellum, from oxidative damage [160][59].
Lipid peroxidation has been related to the initiation and progression of many neurodegenerative disorders, including Alzheimer’s disease (AD), Parkinson’s disease (PD), and amyotrophic lateral sclerosis (ALS) [161][60]. Likewise, PKAN’s pathomechanisms are directly related to the overproduction of ROS and mitochondrial redox imbalance [162][61]. Particularly, lipid peroxidation and increased ROS production have been detected in fibroblast and iNs derived from PKAN’s patients, [41,72][49][62]. Thus, the inhibition of lipid peroxidation propagation might slow the course and ameliorate the severity of PKAN disease.
The positive effects of omega-3 fatty acids treatment in many disorders are now well known by many studies assessing their implication in multiple biochemical functions, including the improvement of antioxidant defenses [163][63], the synthesis of anti-inflammatory factors, increased cellular membranes fluidity, and the modulation of gene expression [164,165,166][64][65][66]. Interestingly, it has been reported that omega-3 fatty acids supplementation also has antioxidant effects by suppressing lipid peroxidation [167][67]. In addition, they have been implicated in synaptic plasticity, contributing to the enhancement of cognitive activity [164][64]. Scientific evidence is accumulating on the potential efficacy of omega-3 fatty acids treatment in neurodegenerative diseases in general [168[68][69],169], and in AD and PD in particular [170][70].
α-Lipoic acid is a pleiotropic organosulfur compound necessary for mitochondrial activity and energy generation, as well as for regulating gene expression [171,172,173][71][72][73]. α-Lipoic acid is produced from plants, animals, and humans and is synthesized de novo in mitochondria using mtFAS II, S-adenosylmethionine, and iron-sulfur group intermediates [173][73]. α-Lipoic acid has a determinant role in oxidative metabolism characterized by its antioxidant properties; this is the reason why it has neuroprotective and anti-inflammatory properties [174][74]. In this respect, α-lipoic acid can decrease the levels of proinflammatory molecules and eliminate ROS and reactive nitrogen species (RNS) [175][75]. In addition, α-lipoic acid supplementation has been shown to reduce lipid peroxidation and increase cellular antioxidant activity [176][76].
From an energetic point of view, α-lipoic acid acts as a cofactor for pyruvate dehydrogenase (PDH), α-ketoglutarate dehydrogenase (KDH), protein H of the glycine cleavage system (GCS) and branched-chain ketoacid dehydrogenase [177,178,179][77][78][79]. Furthermore, several studies have demonstrated that α-lipoic acid also has chelating properties on metals such as iron or copper and a positive impact on oxidative stress and lipid peroxidation [180][80]. These findings suggest that α-lipoic acid is an interesting compound for the treatment of neurodegenerative diseases such as PKAN. Corroborating this hypothesis, α-lipoic acid supplementation decreased significantly iron accumulation in responsive PKAN fibroblasts and iNs [123][39]. These results are also consistent with the positive effect of α-lipoic acid supplementation on reducing the age-dependent iron overload in the rat cerebral cortex [181][81]. Moreover, α-lipoic acid also avoided iron overload caused by ferric ammonium citrate supplementation in a zebrafish model [182][82].
In summary, antioxidants such as vitamin E, omega 3 and α-lipoic acid can protect cell membranes from oxidative stress and lipid peroxidation, a principal pathological feature present in PKAN [19,32][18][83] and other NBIA disorders [117][84].
On the other hand, L-carnitine, a quaternary amine (3-hydroxy-4-N-trimethylaminobutyrate) that is synthesized from the amino acids lysine and methionine, is necessary for the translocation of fatty acids to the mitochondrial compartment for β-oxidation. In addition, L-carnitine has a role in carbohydrate metabolism, stimulates mitochondrial biogenesis by increasing gene expression of mitochondrial components, and prevents the accumulation of toxic products or reactive radicals [183,184][85][86]. Mitochondrial dysfunction in PKAN may impair fatty acid β-oxidation which can preferentially affect brain metabolism. Furthermore, dysfunction of the mitochondrial respiratory chain provokes an increase in the NADH/NAD (+) ratio that inhibits β-oxidation and secondarily L-carnitine deficiency [185][87]. Therefore, L-carnitine as a natural compound that can increase cellular energy production may have therapeutic potential in PKAN. Recently, many works have shown the positive effects of L-carnitine supplementation on mitochondrial function in several pathologies [184,186][86][88].
Furthermore, as PDH deficiency is a major pathologic feature of PKAN, PDH-enhancing agents such as thiamine [187][89] may act as an interesting adjuvant therapy. Thiamine has many functions in cell metabolism since it functions as a cofactor of several multimeric enzymes such as PDH and α-KGDH complexes that participate in the Krebs cycle. In addition, it has been described that thiamine treatment has positive effects in several patients with PDH deficiency due to pyruvate dehydrogenase alpha subunit (E1) mutations [188,189,190,191,192][90][91][92][93][94].
Interestingly, all positive compounds identified after personalized drug screens (pantothenate, pantethine, vitamin E, omega 3, α-lipoic acid, L-carnitine, and thiamine) increased PANK2 transcripts and protein expression levels and up-regulated key transcription factors such as NF-Y, FOXN4, and hnRNPA/B [122,123][38][39] which are involved in PANK2 gene expression [193][95]. Furthermore, it is known that these positive supplements also activate mitochondrial biogenesis through the expression of essential regulators such as peroxisome proliferator-activated receptor coactivator-1α (PGC1α) and mitochondrial transcription factor A TFAM [194,195,196][96][97][98]. Taken together, these data provide useful information on the molecular mechanisms involved in the positive effect of pantothenate, pantethine, vitamin E, α-lipoic acid, omega 3, L-carnitine, and thiamine.
It is hypothesized that partial correction of PANK2 expression levels by these compounds may increase CoA biosynthesis in the mitochondrial compartment, allowing 4′-phosphopantethenylation of essential mitochondrial proteins such as mtACP, mitochondrial10-FTHFDH (ALDH1L2) and AASS [20][99]. In agreement with this hypothesis, the results showed that the expression levels of several 4′-phosphopantetheine carrier proteins in PKAN cells were increased in responsive pathogenic variants after pantothenate, pantethine, vitamin E, omega 3, α-lipoic acid, L-carnitine or thiamine supplementation [122,123][38][39].
3. Polytarget Therapy in PKAN
3. Polytarget Therapy in Pantothenate Kinase-Associated Neurodegeneration
Since several compounds have a positive effect on PKAN cell models, an interesting approach would be to examine their therapeutic efficacy both individually or in combination in controlled clinical trials. In fact, the strategy of combining several compounds that simultaneously affect different cellular pathways or processes are standard procedure in many important therapeutic areas such as cancer, Alzheimer’s disease (AD), Parkinson’s disease (PD), inflammation, epilepsy, depression, and other psychiatric disorders and may be more effective in controlling complex diseases such as PKAN [197,198,199][100][101][102]. Disadvantages of monotherapies can thus be overcome by designing drug combinations that modulate multiple targets [200][103].
Cellular models derived from patients with genetic neurodegenerative diseases allow for the systematic identification of drugs and their potential synergistic combinations that can rapidly move into preclinical development and clinical practice [201,202][104][105].
The progression of neurodegenerative diseases contributes to various factors such as mitochondrial dysfunction, iron accumulation, oxidative stress, inflammation, as well as genetic and environmental factors [203][106]. Therefore, multitargeted therapies with antioxidant and mitochondrial-stimulating compounds may address the multifactorial and complex nature of these diseases more effectively [204,205][107][108]. Multitarget therapeutic approaches have recently become a useful strategy in the development of potential treatments for neurological disorders [206][109].
However, since the crossing of substances to the brain depends on transport mechanisms present in the blood-brain barrier and the diffusion of these compounds also depends on the physicochemical characteristics of the molecule, further studies are warranted on the clinical effects of the positive compounds considering its bioavailability, pharmacokinetics and, in particular, its transport through the blood-brain barrier [207][110].
References
- Hogarth, P.; Kurian, M.A.; Gregory, A.; Csanyi, B.; Zagustin, T.; Kmiec, T.; Wood, P.; Klucken, A.; Scalise, N.; Sofia, F.; et al. Consensus clinical management guideline for pantothenate kinase-associated neurodegeneration (PKAN). Mol. Genet. Metab. 2017, 120, 278–287.
- Gregory, A.; Polster, B.J.; Hayflick, S.J. Clinical and genetic delineation of neurodegeneration with brain iron accumulation. J. Med. Genet. 2009, 46, 73–80.
- Levi, S.; Tiranti, V. Neurodegeneration with Brain Iron Accumulation Disorders: Valuable Models Aimed at Understanding the Pathogenesis of Iron Deposition. Pharmaceuticals 2019, 12, 27.
- Levi, S.; Finazzi, D. Neurodegeneration with brain iron accumulation: Update on pathogenic mechanisms. Front. Pharmacol. 2014, 5, 99.
- Hayflick, S.J. Neurodegeneration with brain iron accumulation: From genes to pathogenesis. Semin. Pediatr. Neurol. 2006, 13, 182–185.
- Hayflick, S.J.; Westaway, S.K.; Levinson, B.; Zhou, B.; Johnson, M.A.; Ching, K.H.; Gitschier, J. Genetic, clinical, and radiographic delineation of Hallervorden-Spatz syndrome. N. Engl. J. Med. 2003, 348, 33–40.
- Leonardi, R.; Zhang, Y.M.; Lykidis, A.; Rock, C.O.; Jackowski, S. Localization and regulation of mouse pantothenate kinase 2. FEBS Lett. 2007, 581, 4639–4644.
- Jackowski, S.; Rock, C.O. CoA regulation and metabolic control. Biochem 2015, 37, 4–8.
- Huang, L.; Khusnutdinova, A.; Nocek, B.; Brown, G.; Xu, X.; Cui, H.; Petit, P.; Flick, R.; Zallot, R.; Balmant, K.; et al. A family of metal-dependent phosphatases implicated in metabolite damage-control. Nat. Chem. Biol. 2016, 12, 621–627.
- Yao, J.; Subramanian, C.; Rock, C.O.; Jackowski, S. Human pantothenate kinase 4 is a pseudo-pantothenate kinase. Protein Sci. 2019, 28, 1031–1047.
- Leonardi, R.; Zhang, Y.M.; Rock, C.O.; Jackowski, S. Coenzyme A: Back in action. Prog. Lipid Res. 2005, 44, 125–153.
- Thakur, N.; Klopstock, T.; Jackowski, S.; Kuscer, E.; Tricta, F.; Videnovic, A.; Jinnah, H.A. Rational Design of Novel Therapies for Pantothenate Kinase-Associated Neurodegeneration. Mov. Disord. 2021, 36, 2005–2016.
- Cavestro, C.; Diodato, D.; Tiranti, V.; Di Meo, I. Inherited Disorders of Coenzyme A Biosynthesis: Models, Mechanisms, and Treatments. Int. J. Mol. Sci. 2023, 24, 5951.
- Munshi, M.I.; Yao, S.J.; Ben Mamoun, C. Redesigning therapies for pantothenate kinase-associated neurodegeneration. J. Biol. Chem. 2022, 298, 101577.
- Hayflick, S.J.; Jeong, S.Y.; Sibon, O.C.M. PKAN pathogenesis and treatment. Mol. Genet. Metab. 2022, 137, 283–291.
- Nixon, R.A. The role of autophagy in neurodegenerative disease. Nat. Med. 2013, 19, 983–997.
- Huang, Y.; Wan, Z.; Tang, Y.; Xu, J.; Laboret, B.; Nallamothu, S.; Yang, C.; Liu, B.; Lu, R.O.; Lu, B.; et al. Pantothenate kinase 2 interacts with PINK1 to regulate mitochondrial quality control via acetyl-CoA metabolism. Nat. Commun. 2022, 13, 2412.
- Alvarez-Cordoba, M.; Fernandez Khoury, A.; Villanueva-Paz, M.; Gomez-Navarro, C.; Villalon-Garcia, I.; Suarez-Rivero, J.M.; Povea-Cabello, S.; de la Mata, M.; Cotan, D.; Talaveron-Rey, M.; et al. Pantothenate Rescues Iron Accumulation in Pantothenate Kinase-Associated Neurodegeneration Depending on the Type of Mutation. Mol. Neurobiol. 2019, 56, 3638–3656.
- Andreotti, G.; Cabeza de Vaca, I.; Poziello, A.; Monti, M.C.; Guallar, V.; Cubellis, M.V. Conformational response to ligand binding in phosphomannomutase2: Insights into inborn glycosylation disorder. J. Biol. Chem. 2014, 289, 34900–34910.
- Goldin, E.; Zheng, W.; Motabar, O.; Southall, N.; Choi, J.H.; Marugan, J.; Austin, C.P.; Sidransky, E. High throughput screening for small molecule therapy for Gaucher disease using patient tissue as the source of mutant glucocerebrosidase. PLoS ONE 2012, 7, e29861.
- Newton, C.L.; Whay, A.M.; McArdle, C.A.; Zhang, M.; van Koppen, C.J.; van de Lagemaat, R.; Segaloff, D.L.; Millar, R.P. Rescue of expression and signaling of human luteinizing hormone G protein-coupled receptor mutants with an allosterically binding small-molecule agonist. Proc. Natl. Acad. Sci. USA 2011, 108, 7172–7176.
- Andreotti, G.; Guarracino, M.R.; Cammisa, M.; Correra, A.; Cubellis, M.V. Prediction of the responsiveness to pharmacological chaperones: Lysosomal human alpha-galactosidase, a case of study. Orphanet J. Rare Dis. 2010, 5, 36.
- Hay Mele, B.; Citro, V.; Andreotti, G.; Cubellis, M.V. Drug repositioning can accelerate discovery of pharmacological chaperones. Orphanet J. Rare Dis. 2015, 10, 55.
- Rigat, B.; Mahuran, D. Diltiazem, a L-type Ca(2+) channel blocker, also acts as a pharmacological chaperone in Gaucher patient cells. Mol. Genet. Metab. 2009, 96, 225–232.
- Maitra, R.; Hamilton, J.W. Altered biogenesis of deltaF508-CFTR following treatment with doxorubicin. Cell Physiol. Biochem. 2007, 20, 465–472.
- Porto, C.; Ferrara, M.C.; Meli, M.; Acampora, E.; Avolio, V.; Rosa, M.; Cobucci-Ponzano, B.; Colombo, G.; Moracci, M.; Andria, G.; et al. Pharmacological enhancement of alpha-glucosidase by the allosteric chaperone N-acetylcysteine. Mol. Ther. 2012, 20, 2201–2211.
- Bendikov-Bar, I.; Maor, G.; Filocamo, M.; Horowitz, M. Ambroxol as a pharmacological chaperone for mutant glucocerebrosidase. Blood Cells Mol. Dis. 2013, 50, 141–145.
- Martin, G.M.; Chen, P.C.; Devaraneni, P.; Shyng, S.L. Pharmacological rescue of trafficking-impaired ATP-sensitive potassium channels. Front. Physiol. 2013, 4, 386.
- Maegawa, G.H.; Tropak, M.; Buttner, J.; Stockley, T.; Kok, F.; Clarke, J.T.; Mahuran, D.J. Pyrimethamine as a potential pharmacological chaperone for late-onset forms of GM2 gangliosidosis. J. Biol. Chem. 2007, 282, 9150–9161.
- Ishihara, K.; Okuyama, S.; Kumano, S.; Iida, K.; Hamana, H.; Murakoshi, M.; Kobayashi, T.; Usami, S.; Ikeda, K.; Haga, Y.; et al. Salicylate restores transport function and anion exchanger activity of missense pendrin mutations. Hear. Res. 2010, 270, 110–118.
- Sharma, L.K.; Subramanian, C.; Yun, M.K.; Frank, M.W.; White, S.W.; Rock, C.O.; Lee, R.E.; Jackowski, S. A therapeutic approach to pantothenate kinase associated neurodegeneration. Nat. Commun. 2018, 9, 4399.
- Strafella, C.; Caputo, V.; Galota, M.R.; Zampatti, S.; Marella, G.; Mauriello, S.; Cascella, R.; Giardina, E. Application of Precision Medicine in Neurodegenerative Diseases. Front. Neurol. 2018, 9, 701.
- Tan, L.; Jiang, T.; Tan, L.; Yu, J.T. Toward precision medicine in neurological diseases. Ann. Transl. Med. 2016, 4, 104.
- Didiasova, M.; Banning, A.; Tikkanen, R. Development of precision therapies for rare inborn errors of metabolism: Functional investigations in cell culture models. J. Inherit. Metab. Dis. 2023, in press.
- Schee Genannt Halfmann, S.; Mahlmann, L.; Leyens, L.; Reumann, M.; Brand, A. Personalized Medicine: What’s in it for Rare Diseases? Adv. Exp. Med. Biol. 2017, 1031, 387–404.
- Kim, J.; Basak, J.M.; Holtzman, D.M. The role of apolipoprotein E in Alzheimer’s disease. Neuron 2009, 63, 287–303.
- Frasier, M.; Fiske, B.K.; Sherer, T.B. Precision medicine for Parkinson’s disease: The subtyping challenge. Front. Aging Neurosci. 2022, 14, 1064057.
- Alvarez-Cordoba, M.; Reche-Lopez, D.; Cilleros-Holgado, P.; Talaveron-Rey, M.; Villalon-Garcia, I.; Povea-Cabello, S.; Suarez-Rivero, J.M.; Suarez-Carrillo, A.; Munuera-Cabeza, M.; Pinero-Perez, R.; et al. Therapeutic approach with commercial supplements for pantothenate kinase-associated neurodegeneration with residual PANK2 expression levels. Orphanet J. Rare Dis. 2022, 17, 311.
- Talaveron-Rey, M.; Alvarez-Cordoba, M.; Villalon-Garcia, I.; Povea-Cabello, S.; Suarez-Rivero, J.M.; Gomez-Fernandez, D.; Romero-Gonzalez, A.; Suarez-Carrillo, A.; Munuera-Cabeza, M.; Cilleros-Holgado, P.; et al. Alpha-lipoic acid supplementation corrects pathological alterations in cellular models of pantothenate kinase-associated neurodegeneration with residual PANK2 expression levels. Orphanet J. Rare Dis. 2023, 18, 80.
- Subramanian, C.; Yun, M.K.; Yao, J.; Sharma, L.K.; Lee, R.E.; White, S.W.; Jackowski, S.; Rock, C.O. Allosteric Regulation of Mammalian Pantothenate Kinase. J. Biol. Chem. 2016, 291, 22302–22314.
- Di Meo, I.; Carecchio, M.; Tiranti, V. Inborn errors of coenzyme A metabolism and neurodegeneration. J. Inherit. Metab. Dis. 2019, 42, 49–56.
- Zano, S.P.; Pate, C.; Frank, M.; Rock, C.O.; Jackowski, S. Correction of a genetic deficiency in pantothenate kinase 1 using phosphopantothenate replacement therapy. Mol. Genet. Metab. 2015, 116, 281–288.
- Balibar, C.J.; Hollis-Symynkywicz, M.F.; Tao, J. Pantethine rescues phosphopantothenoylcysteine synthetase and phosphopantothenoylcysteine decarboxylase deficiency in Escherichia coli but not in Pseudomonas aeruginosa. J. Bacteriol. 2011, 193, 3304–3312.
- Rana, A.; Seinen, E.; Siudeja, K.; Muntendam, R.; Srinivasan, B.; van der Want, J.J.; Hayflick, S.; Reijngoud, D.J.; Kayser, O.; Sibon, O.C. Pantethine rescues a Drosophila model for pantothenate kinase-associated neurodegeneration. Proc. Natl. Acad. Sci. USA 2010, 107, 6988–6993.
- Zizioli, D.; Tiso, N.; Guglielmi, A.; Saraceno, C.; Busolin, G.; Giuliani, R.; Khatri, D.; Monti, E.; Borsani, G.; Argenton, F.; et al. Knock-down of pantothenate kinase 2 severely affects the development of the nervous and vascular system in zebrafish, providing new insights into PKAN disease. Neurobiol. Dis. 2016, 85, 35–48.
- Brunetti, D.; Dusi, S.; Giordano, C.; Lamperti, C.; Morbin, M.; Fugnanesi, V.; Marchet, S.; Fagiolari, G.; Sibon, O.; Moggio, M.; et al. Pantethine treatment is effective in recovering the disease phenotype induced by ketogenic diet in a pantothenate kinase-associated neurodegeneration mouse model. Brain 2014, 137, 57–68.
- Evans, M.; Rumberger, J.A.; Azumano, I.; Napolitano, J.J.; Citrolo, D.; Kamiya, T. Pantethine, a derivative of vitamin B5, favorably alters total, LDL and non-HDL cholesterol in low to moderate cardiovascular risk subjects eligible for statin therapy: A triple-blinded placebo and diet-controlled investigation. Vasc. Health Risk Manag. 2014, 10, 89–100.
- Chang, X.; Zhang, J.; Jiang, Y.; Yao, B.; Wang, J.; Wu, Y. Pilot trial on the efficacy and safety of pantethine in children with pantothenate kinase-associated neurodegeneration: A single-arm, open-label study. Orphanet J. Rare Dis. 2020, 15, 248.
- Campanella, A.; Privitera, D.; Guaraldo, M.; Rovelli, E.; Barzaghi, C.; Garavaglia, B.; Santambrogio, P.; Cozzi, A.; Levi, S. Skin fibroblasts from pantothenate kinase-associated neurodegeneration patients show altered cellular oxidative status and have defective iron-handling properties. Hum. Mol. Genet. 2012, 21, 4049–4059.
- Kotzbauer, P.T.; Truax, A.C.; Trojanowski, J.Q.; Lee, V.M. Altered neuronal mitochondrial coenzyme A synthesis in neurodegeneration with brain iron accumulation caused by abnormal processing, stability, and catalytic activity of mutant pantothenate kinase 2. J. Neurosci. 2005, 25, 689–698.
- Girotti, A.W. Lipid hydroperoxide generation, turnover, and effector action in biological systems. J. Lipid Res. 1998, 39, 1529–1542.
- Yin, H.; Xu, L.; Porter, N.A. Free radical lipid peroxidation: Mechanisms and analysis. Chem. Rev. 2011, 111, 5944–5972.
- Van der Paal, J.; Neyts, E.C.; Verlackt, C.C.W.; Bogaerts, A. Effect of lipid peroxidation on membrane permeability of cancer and normal cells subjected to oxidative stress. Chem. Sci. 2016, 7, 489–498.
- Ayala, A.; Munoz, M.F.; Arguelles, S. Lipid peroxidation: Production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxidative Med. Cell. Longev. 2014, 2014, 360438.
- Villalon-Garcia, I.; Povea-Cabello, S.; Alvarez-Cordoba, M.; Talaveron-Rey, M.; Suarez-Rivero, J.M.; Suarez-Carrillo, A.; Munuera-Cabeza, M.; Reche-Lopez, D.; Cilleros-Holgado, P.; Pinero-Perez, R.; et al. Vicious cycle of lipid peroxidation and iron accumulation in neurodegeneration. Neural Regen. Res. 2023, 18, 1196–1202.
- Burton, G.W.; Joyce, A.; Ingold, K.U. First proof that vitamin E is major lipid-soluble, chain-breaking antioxidant in human blood plasma. Lancet 1982, 2, 327.
- Traber, M.G. Vitamin E: Necessary nutrient for neural development and cognitive function. Proc. Nutr. Soc. 2021, 80, 319–326.
- Ricciarelli, R.; Argellati, F.; Pronzato, M.A.; Domenicotti, C. Vitamin E and neurodegenerative diseases. Mol. Asp. Med. 2007, 28, 591–606.
- Ulatowski, L.M.; Manor, D. Vitamin E and neurodegeneration. Neurobiol. Dis. 2015, 84, 78–83.
- Pena-Bautista, C.; Vento, M.; Baquero, M.; Chafer-Pericas, C. Lipid peroxidation in neurodegeneration. Clin. Chim. Acta Int. J. Clin. Chem. 2019, 497, 178–188.
- Espinos, C.; Galindo, M.I.; Garcia-Gimeno, M.A.; Ibanez-Cabellos, J.S.; Martinez-Rubio, D.; Millan, J.M.; Rodrigo, R.; Sanz, P.; Seco-Cervera, M.; Sevilla, T.; et al. Oxidative Stress, a Crossroad Between Rare Diseases and Neurodegeneration. Antioxidants 2020, 9, 313.
- Orellana, D.I.; Santambrogio, P.; Rubio, A.; Yekhlef, L.; Cancellieri, C.; Dusi, S.; Giannelli, S.G.; Venco, P.; Mazzara, P.G.; Cozzi, A.; et al. Coenzyme A corrects pathological defects in human neurons of PANK2-associated neurodegeneration. EMBO Mol. Med. 2016, 8, 1197–1211.
- Heshmati, J.; Morvaridzadeh, M.; Maroufizadeh, S.; Akbari, A.; Yavari, M.; Amirinejad, A.; Maleki-Hajiagha, A.; Sepidarkish, M. Omega-3 fatty acids supplementation and oxidative stress parameters: A systematic review and meta-analysis of clinical trials. Pharmacol. Res. 2019, 149, 104462.
- Avallone, R.; Vitale, G.; Bertolotti, M. Omega-3 Fatty Acids and Neurodegenerative Diseases: New Evidence in Clinical Trials. Int. J. Mol. Sci. 2019, 20, 4256.
- Calon, F.; Cole, G. Neuroprotective action of omega-3 polyunsaturated fatty acids against neurodegenerative diseases: Evidence from animal studies. Prostaglandins Leukot. Essent. Fat. Acids 2007, 77, 287–293.
- Eckert, G.P.; Lipka, U.; Muller, W.E. Omega-3 fatty acids in neurodegenerative diseases: Focus on mitochondria. Prostaglandins Leukot. Essent. Fat. Acids 2013, 88, 105–114.
- Da Silva, E.P., Jr.; Nachbar, R.T.; Levada-Pires, A.C.; Hirabara, S.M.; Lambertucci, R.H. Omega-3 fatty acids differentially modulate enzymatic anti-oxidant systems in skeletal muscle cells. Cell Stress. Chaperones 2016, 21, 87–95.
- Calviello, G.; Su, H.M.; Weylandt, K.H.; Fasano, E.; Serini, S.; Cittadini, A. Experimental evidence of omega-3 polyunsaturated fatty acid modulation of inflammatory cytokines and bioactive lipid mediators: Their potential role in inflammatory, neurodegenerative, and neoplastic diseases. Biomed. Res. Int. 2013, 2013, 743171.
- Cardoso, C.; Afonso, C.; Bandarra, N.M. Dietary DHA and health: Cognitive function ageing. Nutr. Res. Rev. 2016, 29, 281–294.
- Moore, K.; Hughes, C.F.; Ward, M.; Hoey, L.; McNulty, H. Diet, nutrition and the ageing brain: Current evidence and new directions. Proc. Nutr. Soc. 2018, 77, 152–163.
- Gomes, M.B.; Negrato, C.A. Alpha-lipoic acid as a pleiotropic compound with potential therapeutic use in diabetes and other chronic diseases. Diabetol. Metab. Syndr. 2014, 6, 80.
- Salehi, B.; Berkay Yilmaz, Y.; Antika, G.; Boyunegmez Tumer, T.; Fawzi Mahomoodally, M.; Lobine, D.; Akram, M.; Riaz, M.; Capanoglu, E.; Sharopov, F.; et al. Insights on the Use of alpha-Lipoic Acid for Therapeutic Purposes. Biomolecules 2019, 9, 356.
- Solmonson, A.; DeBerardinis, R.J. Lipoic acid metabolism and mitochondrial redox regulation. J. Biol. Chem. 2018, 293, 7522–7530.
- Moura, F.A.; de Andrade, K.Q.; dos Santos, J.C.; Goulart, M.O. Lipoic Acid: Its antioxidant and anti-inflammatory role and clinical applications. Curr. Top. Med. Chem. 2015, 15, 458–483.
- Tibullo, D.; Li Volti, G.; Giallongo, C.; Grasso, S.; Tomassoni, D.; Anfuso, C.D.; Lupo, G.; Amenta, F.; Avola, R.; Bramanti, V. Biochemical and clinical relevance of alpha lipoic acid: Antioxidant and anti-inflammatory activity, molecular pathways and therapeutic potential. Inflamm. Res. 2017, 66, 947–959.
- Molz, P.; Schroder, N. Potential Therapeutic Effects of Lipoic Acid on Memory Deficits Related to Aging and Neurodegeneration. Front. Pharmacol. 2017, 8, 849.
- Dos Santos, S.M.; Romeiro, C.F.R.; Rodrigues, C.A.; Cerqueira, A.R.L.; Monteiro, M.C. Mitochondrial Dysfunction and Alpha-Lipoic Acid: Beneficial or Harmful in Alzheimer’s Disease? Oxidative Med. Cell. Longev. 2019, 2019, 8409329.
- Perham, R.N. Swinging arms and swinging domains in multifunctional enzymes: Catalytic machines for multistep reactions. Annu. Rev. Biochem. 2000, 69, 961–1004.
- Smith, A.R.; Shenvi, S.V.; Widlansky, M.; Suh, J.H.; Hagen, T.M. Lipoic acid as a potential therapy for chronic diseases associated with oxidative stress. Curr. Med. Chem. 2004, 11, 1135–1146.
- Rezaei Zonooz, S.; Hasani, M.; Morvaridzadeh, M.; Beatriz Pizarro, A.; Heydari, H.; Yosaee, S.; Rezamand, G.; Heshmati, J. Effect of alpha-lipoic acid on oxidative stress parameters: A systematic review and meta-analysis. J. Funct. Foods 2021, 87, 104774.
- Suh, J.H.; Moreau, R.; Heath, S.H.; Hagen, T.M. Dietary supplementation with (R)-alpha-lipoic acid reverses the age-related accumulation of iron and depletion of antioxidants in the rat cerebral cortex. Redox Rep. 2005, 10, 52–60.
- Camiolo, G.; Tibullo, D.; Giallongo, C.; Romano, A.; Parrinello, N.L.; Musumeci, G.; Di Rosa, M.; Vicario, N.; Brundo, M.V.; Amenta, F.; et al. alpha-Lipoic Acid Reduces Iron-induced Toxicity and Oxidative Stress in a Model of Iron Overload. Int. J. Mol. Sci. 2019, 20, 609.
- Alvarez-Cordoba, M.; Talaveron-Rey, M.; Villalon-Garcia, I.; Povea-Cabello, S.; Suarez-Rivero, J.M.; Suarez-Carrillo, A.; Munuera-Cabeza, M.; Salas, J.J.; Sanchez-Alcazar, J.A. Down regulation of the expression of mitochondrial phosphopantetheinyl-proteins in pantothenate kinase-associated neurodegeneration: Pathophysiological consequences and therapeutic perspectives. Orphanet J. Rare Dis. 2021, 16, 201.
- Villalon-Garcia, I.; Alvarez-Cordoba, M.; Povea-Cabello, S.; Talaveron-Rey, M.; Villanueva-Paz, M.; Luzon-Hidalgo, R.; Suarez-Rivero, J.M.; Suarez-Carrillo, A.; Munuera-Cabeza, M.; Salas, J.J.; et al. Vitamin E prevents lipid peroxidation and iron accumulation in PLA2G6-Associated Neurodegeneration. Neurobiol. Dis. 2022, 165, 105649.
- Liufu, T.; Wang, Z. Treatment for mitochondrial diseases. Rev. Neurosci. 2021, 32, 35–47.
- Modanloo, M.; Shokrzadeh, M. Analyzing Mitochondrial Dysfunction, Oxidative Stress, and Apoptosis: Potential Role of L-carnitine. Iran. J. Kidney Dis. 2019, 13, 74–86.
- Infante, J.P.; Huszagh, V.A. Secondary carnitine deficiency and impaired docosahexaenoic (22:6n-3) acid synthesis: A common denominator in the pathophysiology of diseases of oxidative phosphorylation and beta-oxidation. FEBS Lett. 2000, 468, 1–5.
- Mantle, D.; Hargreaves, I.P. Mitochondrial Dysfunction and Neurodegenerative Disorders: Role of Nutritional Supplementation. Int. J. Mol. Sci. 2022, 23, 2603.
- Lonsdale, D. A review of the biochemistry, metabolism and clinical benefits of thiamin(e) and its derivatives. Evid. Based Complement. Altern. Med. 2006, 3, 49–59.
- Marsac, C.; Benelli, C.; Desguerre, I.; Diry, M.; Fouque, F.; De Meirleir, L.; Ponsot, G.; Seneca, S.; Poggi, F.; Saudubray, J.M.; et al. Biochemical and genetic studies of four patients with pyruvate dehydrogenase E1 alpha deficiency. Hum. Genet. 1997, 99, 785–792.
- Naito, E.; Ito, M.; Takeda, E.; Yokota, I.; Yoshijima, S.; Kuroda, Y. Molecular analysis of abnormal pyruvate dehydrogenase in a patient with thiamine-responsive congenital lactic acidemia. Pediatr. Res. 1994, 36, 340–346.
- Naito, E.; Ito, M.; Yokota, I.; Saijo, T.; Chen, S.; Maehara, M.; Kuroda, Y. Concomitant administration of sodium dichloroacetate and thiamine in west syndrome caused by thiamine-responsive pyruvate dehydrogenase complex deficiency. J. Neurol. Sci. 1999, 171, 56–59.
- Naito, E.; Ito, M.; Yokota, I.; Saijo, T.; Matsuda, J.; Ogawa, Y.; Kitamura, S.; Takada, E.; Horii, Y.; Kuroda, Y. Thiamine-responsive pyruvate dehydrogenase deficiency in two patients caused by a point mutation (F205L and L216F) within the thiamine pyrophosphate binding region. Biochim. Biophys. Acta 2002, 1588, 79–84.
- Naito, E.; Ito, M.; Yokota, I.; Saijo, T.; Matsuda, J.; Osaka, H.; Kimura, S.; Kuroda, Y. Biochemical and molecular analysis of an X-linked case of Leigh syndrome associated with thiamin-responsive pyruvate dehydrogenase deficiency. J. Inherit. Metab. Dis. 1997, 20, 539–548.
- Polster, B.J.; Yoon, M.Y.; Hayflick, S.J. Characterization of the human PANK2 promoter. Gene 2010, 465, 53–60.
- Ekstrand, M.I.; Falkenberg, M.; Rantanen, A.; Park, C.B.; Gaspari, M.; Hultenby, K.; Rustin, P.; Gustafsson, C.M.; Larsson, N.G. Mitochondrial transcription factor A regulates mtDNA copy number in mammals. Hum. Mol. Genet. 2004, 13, 935–944.
- Puigserver, P.; Spiegelman, B.M. Peroxisome proliferator-activated receptor-gamma coactivator 1 alpha (PGC-1 alpha): Transcriptional coactivator and metabolic regulator. Endocr. Rev. 2003, 24, 78–90.
- Wu, Z.; Puigserver, P.; Andersson, U.; Zhang, C.; Adelmant, G.; Mootha, V.; Troy, A.; Cinti, S.; Lowell, B.; Scarpulla, R.C.; et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 1999, 98, 115–124.
- Lambrechts, R.A.; Schepers, H.; Yu, Y.; van der Zwaag, M.; Autio, K.J.; Vieira-Lara, M.A.; Bakker, B.M.; Tijssen, M.A.; Hayflick, S.J.; Grzeschik, N.A.; et al. CoA-dependent activation of mitochondrial acyl carrier protein links four neurodegenerative diseases. EMBO Mol. Med. 2019, 11, e10488.
- Talevi, A. Multi-target pharmacology: Possibilities and limitations of the “skeleton key approach” from a medicinal chemist perspective. Front. Pharmacol. 2015, 6, 205.
- Xu, Y.; Li, X.J. Multi-target therapeutics and new drug discovery. Yao Xue Xue Bao 2009, 44, 226–230.
- Zimmermann, G.R.; Lehar, J.; Keith, C.T. Multi-target therapeutics: When the whole is greater than the sum of the parts. Drug Discov. Today 2007, 12, 34–42.
- Keith, C.T.; Borisy, A.A.; Stockwell, B.R. Multicomponent therapeutics for networked systems. Nat. Rev. Drug Discov. 2005, 4, 71–78.
- Borisy, A.A.; Elliott, P.J.; Hurst, N.W.; Lee, M.S.; Lehar, J.; Price, E.R.; Serbedzija, G.; Zimmermann, G.R.; Foley, M.A.; Stockwell, B.R.; et al. Systematic discovery of multicomponent therapeutics. Proc. Natl. Acad. Sci. USA 2003, 100, 7977–7982.
- Butcher, E.C. Can cell systems biology rescue drug discovery? Nat. Rev. Drug Discov. 2005, 4, 461–467.
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795.
- Ibrahim, M.M.; Gabr, M.T. Multitarget therapeutic strategies for Alzheimer’s disease. Neural Regen. Res. 2019, 14, 437–440.
- Maramai, S.; Benchekroun, M.; Gabr, M.T.; Yahiaoui, S. Multitarget Therapeutic Strategies for Alzheimer’s Disease: Review on Emerging Target Combinations. Biomed. Res. Int. 2020, 2020, 5120230.
- Bawa, P.; Pradeep, P.; Kumar, P.; Choonara, Y.E.; Modi, G.; Pillay, V. Multi-target therapeutics for neuropsychiatric and neurodegenerative disorders. Drug Discov. Today 2016, 21, 1886–1914.
- Jackowski, S. Proposed Therapies for Pantothenate-Kinase-Associated Neurodegeneration. J. Exp. Neurosci. 2019, 13, 1179069519851118.
More