Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Rita Xu and Version 3 by Rita Xu.
Cellular senescence, a state of permanent cell cycle arrest in response to endogenous and exogenous stimuli, triggers a series of gradual alterations in structure, metabolism, and function, as well as inflammatory gene expression that nurtures a low-grade proinflammatory milieu in human tissue. A growing body of evidence indicates an accumulation of senescent neurons and blood vessels in response to stress and aging in the retina. Prolonged accumulation of senescent cells and long-term activation of stress signaling responses may lead to multiple chronic diseases, tissue dysfunction, and age-related pathologies by exposing neighboring cells to the heightened pathological senescence-associated secretory phenotype (SASP).
- cellular senescence
- senescence-associated secretory phenotype (SASP)
- retinal vascular system
1. Cellular Senescence
1.1. Cell Senescence and Inducers
Throughout life, cells are constantly challenged by internal and external stressors. Repair, cell death, and senescence are the main physiological programs adopted by tissue cells to respond to triggers, depending on the severity and nature of stress. Cellular senescence is a state of irreversible cell cycle arrest, with a preserved metabolic function, that is stimulated in normal cells in response to various internal or external stress/stimuli, as well as developmental signals [1]. The first evidence of senescence was introduced in 1961 when Leonard Hayflick and Paul Moorhead discovered limited proliferation capacity in human fibroblasts after extensive serial passaging [2]. According to the nature of stimuli, cellular senescence is classified into two categories: replicative and stress-induced senescence. Replicative senescence (RS) is triggered by telomere attrition, whereas stress-induced premature senescence (SIPS) is caused by stressors such as unresolved DNA damage, oxidative stress, activated oncogenes, irradiation, mitochondrial dysfunction, genotoxic drugs, cell–cell fusion, epigenetic modifiers, and impaired proteostasis [3]. An intriguing aspect of cellular senescence is that although characteristically occurring in injured tissues, in which it is transient and important for tissue regeneration, it increases with aging and happens during embryonic development. Thus, cellular senescence is found in homeostatic biological processes of very distinct etiology.
1.2. Cell Cycle Arrest
An active cell cycle in multicellular eukaryotes is divided into four distinct phases: G1 (a decision window to enter or exit the cell cycle), S (DNA duplication), G2 (a decision window to initiate or cease the process that leads to chromosome segregation), and M (DNA segregation) [4]. Cell cycle progression is driven by phase-specific cyclin-CDK complexes that phosphorylate important regulatory targets. In the pre-replicative G1 phase, the accumulation of cyclin D-CDK4/6 complexes commits cells to enter the next cell cycle, thereby inhibiting cell cycle exit (G0 state) [4]. Indeed, there is a decision window here that allows cells to continue or exit the cell cycle transiently or permanently. Subsequent accumulation of cyclin E-CDK2 inhibits retinoblastoma protein (RB), leading to cyclin A-CDK2 accumulation, replication initiation, and S phase entry [4]. After S phase completion, the entry into mitosis and the -mediated degradation of cyclins, which is necessary to complete the cell cycle, are driven by the activity of the cyclin A/B-CDK1 complex [4].
Cell cycle arrest, regardless of the cell type, is the primary feature of senescent cells. However, growth cessation is not an exclusive characteristic of senescent cells, being also observed in cellular quiescence and terminally differentiated cells.
Terminally differentiated cells: The development, maintenance, and regeneration of many human tissues highly depend on the terminal differentiation of resident progenitor cells [5]. During terminal differentiation, undifferentiated progenitor cells undergo a gradual process of specialization and permanently exit the cell cycle upon reaching a fully differentiated post-mitotic state [6]. These highly specialized cells are refractory to pro-mitogenic signals and work together to facilitate the specialized functions of a tissue. Previous studies indicate that cells can undergo terminal differentiation and exit the cell cycle at G1 simultaneously, and this transformation is made possible by the intricate cooperation of multiple molecules, including CIP/KIP inhibitors, pRB, and [5][6]. In contrast to other non-proliferating cells, such as those in quiescence and senescence, the morphology, metabolic rate, and transcriptome profile of terminally differentiated cells are significantly shaped by their typical cellular functions within a specific tissue. Neurons, muscles, adipocytes, and bone cells are some examples of terminally differentiated cells that exit the cell cycle permanently while adopting cell type-specific transcriptional programs [6].
Cellular quiescence: Cell cycle arrest in quiescent cells is reversible, as it enables cells to return to the normal cell cycle in response to certain pro-mitogenic factors and signals. For instance, tissue injury stimulates local resident quiescent adult stem cells (qASCs) to re-enter the cell cycle, causing the production of transient progenitor and mature functional cells that promote repair and tissue regeneration [7]. In general, proliferation arrest in quiescent cells is mediated by cyclin-dependent kinase inhibitors (CDKIs) including , and at G0 [8][9]. Nonetheless, quiescent cells are not only restricted in terms of replication ability, but are also characterized by a reduced size, dense heterochromatin, low metabolic activity, and reversible suppression of global RNA and protein synthesis [10]. Quiescent cells satisfy their lower energy demand by producing ATP via glycolytic pathways and fatty acid oxidation, and therefore have lower oxidative phosphorylation and mitochondrial activity [10].
1.3. The Senescent Phenotype
Cell senescence is associated with dramatic changes in cell morphology, structure, and metabolism (Figure 1). The flattened, enlarged, and multinucleated morphology of senescent cells (SCs) reflects changes occurring in the number and function of membranous organelles such as lysosomes, mitochondria, endoplasmic reticulum (ER), nucleus, etc. Senescent cells, both in vivo and in vitro, are often characterized by multiple nuclei within an enlarged cytoplasm [11][12]. It has been proposed that multinucleated senescent cells can be generated through endomitosis/cytokinesis failure [13][14][15], cell–cell fusion [13], and a process referred to as “amitosis” (involving the fragmentation of polyploid nuclei during interphase) [16]. However, a previously published study by Dikovskaya et al. proposed a mechanism indicating that multinucleation in senescent cells occurs predominantly due to mitotic failure [17].
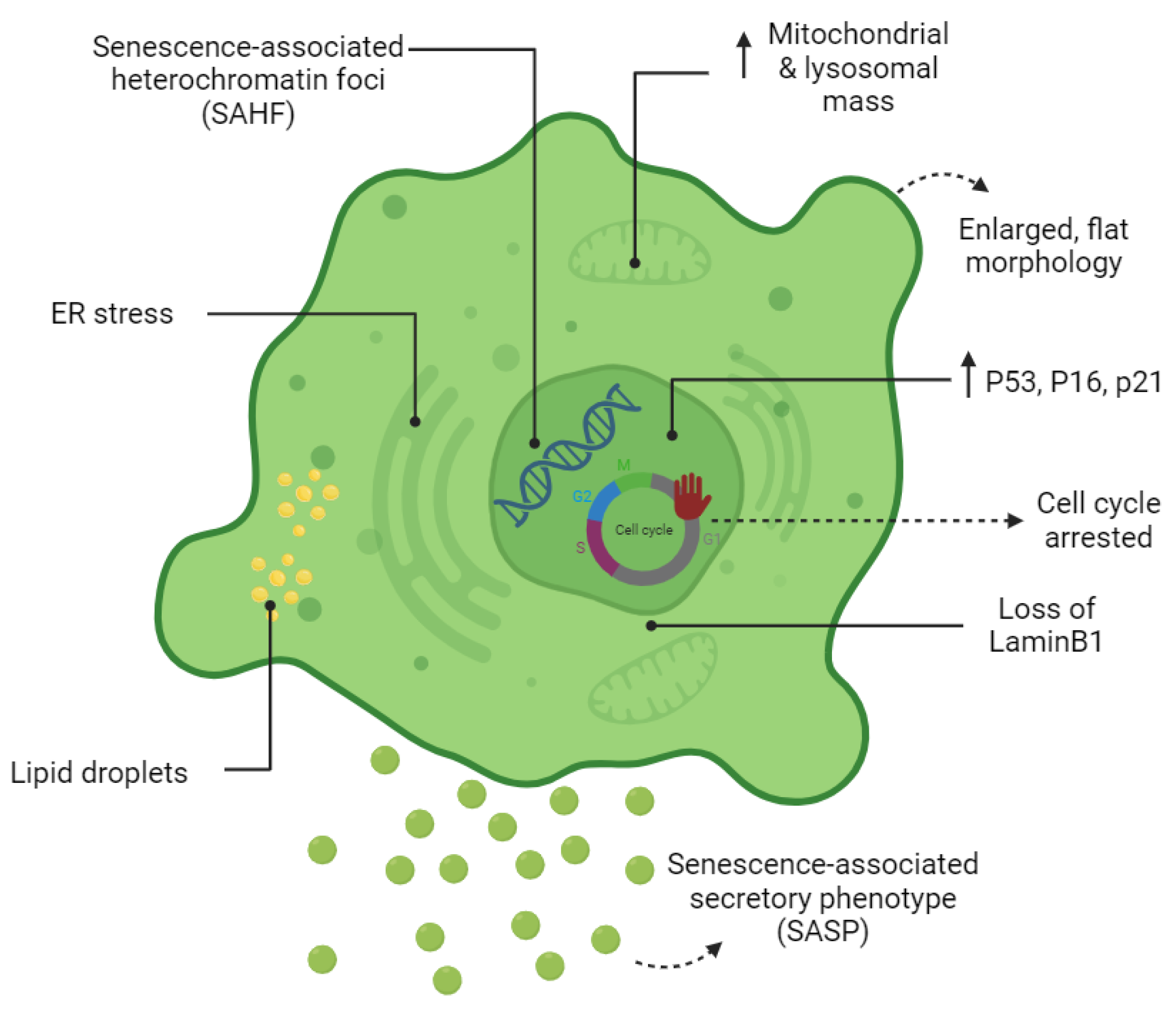
Figure 1. Cardinal features of senescent cells. The morphology, metabolism, and biomarkers of senescent cells are illustrated.
Increased lysosomal biogenesis and elevated autophagic activity are documented during the establishment of SCs [18]. Furthermore, the protein compositions of lysosomes are shown to change significantly upon senescence [18]. Lysosomal hydrolytic enzymes are required for the degradation of damaged organelles and proteins via the autophagic pathway. Lysosomal β-galactosidase is the most widely used biomarker for SCs and becomes detectable at a pH of 6.0 due to the increased lysosomal mass in affected cells [19]. However, the drawback of using this marker as a reporter of senescence is that SA-β-gal activity is also detected in non-senescent cells cultured under confluent or serum-starved conditions [20]. Lipofuscin is a complex fluorescent mixture of highly oxidized proteins and lipids that due to their cross-linked nature are non-degradable and accumulate in lysosomes, only being segregated during cell division [21][22]. Lipofuscin accumulation, which can be detected by Sudan Black B staining, is another hallmark of SCs.
Although the cell cycle is arrested in SCs, these cells are still metabolically active. At a senescent state, cells have higher metabolic demands due to their enlarged size and to their robust production of secreted proteins (SASP) [23]. Generally, cells produce units of energy (ATP) through mitochondrial oxidative phosphorylation (OXPHOS) and anaerobic glycolysis in the presence and absence of oxygen, respectively [24]. It has become apparent that during cellular senescence, the metabolism shifts from oxidative phosphorylation (OXPHOS) towards glycolysis even in presence of high oxygen levels [25][26][27]. Mitochondrial mass increases in SCs may partially be a consequence of the increased size of cells in senescence [28]. However, decreased mitophagy activity in senescence promotes the accumulation of dysfunctional mitochondria in SCs [28]. Although mitochondrial mass increases dramatically in SCs, they are characterized by a reduced respiratory capacity [28]. Reactive oxygen species (ROS) are a by-product of mitochondrial respiration and increase strikingly in SCs due to low mitochondrial membrane potential [28][29]. Mitochondria-derived ROS cause mitochondrial damage and DNA breaks at the telomeric regions leading to cell cycle arrest [29][30]. It has also been shown that cytosolic p53 accumulation in SCs concurrent to ROS generation in SCs results in Parkin inhibition (an important regulator of mitophagy) and mitochondrial dysfunction [31]. Generally, NADH molecules are oxidized in the mitochondria to NAD+. In mitochondrial dysfunction-associated senescence (MiDAS), accumulation of cytosolic NADH results in the inhibition of glycolytic enzymes, ATP depletion, and finally, cell cycle arrest [32].
Upregulation of SASP genes is one of the most important phenotypic changes occurring in SCs (Figure 1). The synthesis of SASP factors, which include a group of proinflammatory cytokines, chemokines, proteases, and growth factors, results in ER expansion in SCs. Excessive protein synthesis in the ER leads to the accumulation of misfolded proteins and activates the unfolded protein response (UPR) [33]. The ATF6α pathway of UPR is particularly responsible for increasing the size of the ER in SCs [34][35]. The chromatin structure of SCs also changes globally by the formation of senescence-associated heterochromatin foci (SAHF); the latter are specialized regions of facultative heterochromatin that suppress transcription of proliferation-promoting genes [36]. SAHF structures can be detected by DAPI and by antibodies specific to components of SAHF such as H3K9Me2/3, macroH2A, and HP1 proteins. However, SAHF are not a universal biomarker and appear to be induced in SCs by activated oncogenes and DNA replication stressors [37].
1.4. Senescence Molecular Pathways
During cellular senescence, growth arrest occurs through the activation of both the p53/p21(CIP1/WAF1) and p16INK4a/RB signaling pathways (Figure 2). In the p53-dependent senescence pathway, telomere attrition or elevated ROS levels, generated by intrinsic and extrinsic stimuli such as genotoxic stress and mitochondrial dysfunction, result in DNA damage and activation of the DNA damage response (DDR) pathway [38]. The DDR network involves different regulatory proteins including ATM/ATR, CHK1/CHK2, p53, and p21(CIP/WAF1). ATR/CHK1 and ATM/CHK2 signaling are usually activated by single-strand and double-strand DNA breaks (DSBs), respectively [39][40][41]. p53 phosphorylation is induced in both pathways, which allows its binding to the CDKN1A promoter and upregulation of p21(CIP/WAF1) expression. The latter induces senescence by inhibiting the kinase activity of cyclin-CDK complexes and RB phosphorylation [42]. In the p53-independent senescence pathway, the CDK4/6-CyclinD complexes are directly inhibited by p16INK4a and lose their regulatory effects on RB phosphorylation (Figure 2). Dephosphorylated RB binds to the E2F and induces cellular senescence [43].
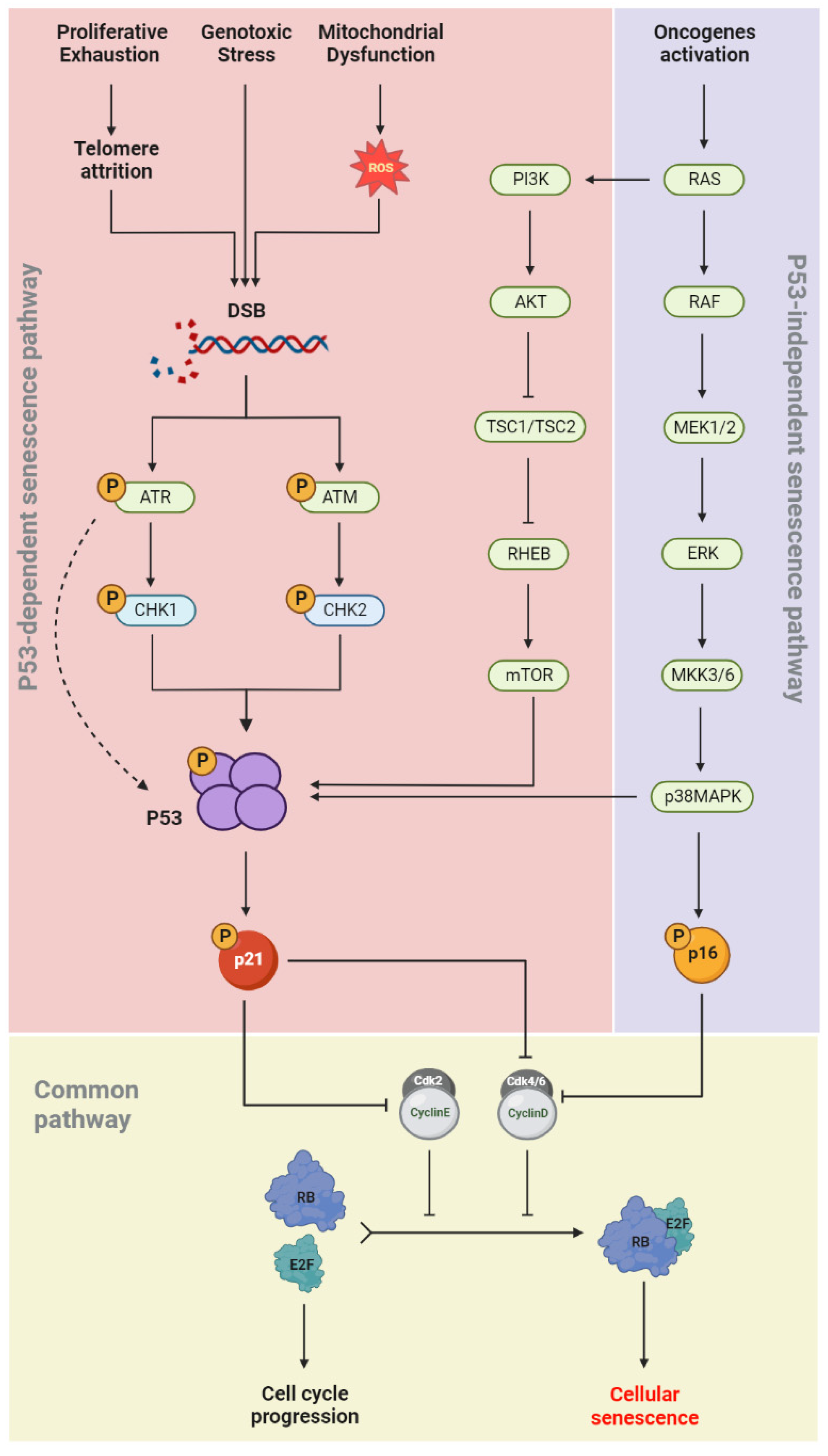
Figure 2. Senescence molecular pathways. Cell cycle arrest occurs in senescent cells via two independent pathways: p53-dependet senescence pathway and p53-independent senescence pathway. Senescence inducers and the main effective downstream molecules are illustrated.
1.5. Autophagy: A Pro-Senescence or an Anti-Senescence Mechanism?
Cellular autophagy is known to decrease with aging, while SCs accumulate in tissues with advancing age [44]. Autophagy was initially considered a mechanism preventing cell senescence, through the elimination of damaged components (e.g., proteins or mitochondria) [45]. This assumption is supported by several studies demonstrating the anti-senescence effects of autophagy. For example, the re-establishment of basal autophagy activity in aged satellite cells prevents cellular senescence and restores the regenerative capacity of geriatric satellite cells [46][47]. Moreover, Kang et al. found that autophagy impairment in removing defective mitochondria induces cellular senescence in primary human fibroblasts in an ROS- and p53-dependent manner [48]. However, this overriding view has been challenged by several recently studies. Targeting essential genes for autophagy, ATG5 and ATG7 delayed the senescence state and reduced the production of the key SASP factors including IL-6 and IL-8 [49]. Further research has also underlined the contribution of a specialized type of autophagy called the TOR-autophagy spatial coupling compartment (TASCC), in the massive synthesis of the SASP factors during the establishment of oncogene-induced senescence (OIS) [50]. Additional evidence for the pro-senescence effects of autophagy has been provided by Nam et al. whose results demonstrated that prolonged activation of autophagy via mTOR inhibitors induces cellular senescence in radiation-resistant cancer cells [51]. These discrepancies may be explained by the type of autophagy involved (basal or induced), as well as by when and where autophagy acts [44][45]. However, the complex interplay between autophagy and cell senescence still needs further investigation to clarify the context-dependent implications of autophagy in cellular senescence.
2. Implications of SCs on the Retinal Vascular System
The retina is one of the most energy-demanding tissues in the human body, and its structural and functional integrity chiefly depends on a regular oxygen/nutrient supply [52][53]. Dysregulated angiogenesis and sustained imbalances in the supply of nutrients and oxygen stimulate stress responses in vital tissues of the retina such as cellular senescence [54]. Upregulation of classical senescence-associated markers including p53, p16INK4a, plasminogen activator inhibitor 1 (Pai1), Cdkn1α (p21(CIP/WAF1)), and promyelocytic leukemia protein (PML) provide further evidence for the presence of senescent retinal cells in retinopathies [53]. However, the response of nervous and vascular cells to the hypoxic/oxidative nature of ischemic retinopathy is not homogeneous across all retinal layers. At P14, cellular senescence is confined to avascular regions and then spreads to pathological vascular tufts and retinal microglia [53]. Apoptotic cell death occurs more frequently in cells of the inner nuclear layer (INL); however, cells of the RGC layer (GCL) adopt a senescent phenotype [53].2.1. Detrimental Effect: Vascular Degeneration
Retina blood vessel is made up of three distinct layers of the tunica intima, the tunica media, and the tunica adventitia. The tunica adventitia consists of fibroblast cells, while the tunica media is hosting smooth muscle cells. The tunica intima, the thinnest layer of the vascular wall, is composed of one layer of endothelial cells (ECs) and a basement membrane. ECSs not only act as a physical barrier but also participate in the regulation of vascular homeostasis, vessel tone, inflammatory responses, and neovascularization [55]. A previous study by Bertelli et al. demonstrated that long-term high glucose exposure stimulates premature senescence in human retinal ECs [56]. Senescent ECs displayed limited replicative potential and increased susceptibility to pathological attacks [57]. Losing replicative potential inhibits cell endothelialization and declines the repair function of vessels [57]. The presence and accumulation of senescent vascular cells also results in a leaky and highly permeable endothelium, allowing immune cells and circulating non-immune cells to enter the surrounding tissue and provoke a low-grade inflammatory state [58][59][60]. In both in vivo and in vitro studies of the blood-brain barrier (BBB) model, the accumulation of senescent vascular cells is accompanied by lower barrier integrity [61]. A study by Venkatesh et al. also demonstrated that EC senescence is associated with increased permeability due to the alteration of vascular endothelial (VE)-cadherin and β-catenin expression and localization [62]. Krouwer et al. provided further evidence for the detrimental effect of senescent ECs where the presence of replicative senescence in a non-senescent endothelial monolayer impaired both adherence and tight junctions and compromised the integrity of the endothelial barrier [63]. Altogether, these studies indicate that the senescence phenotype of ECs is associated with lower barrier integrity in retinal blood vessels (Figure 3).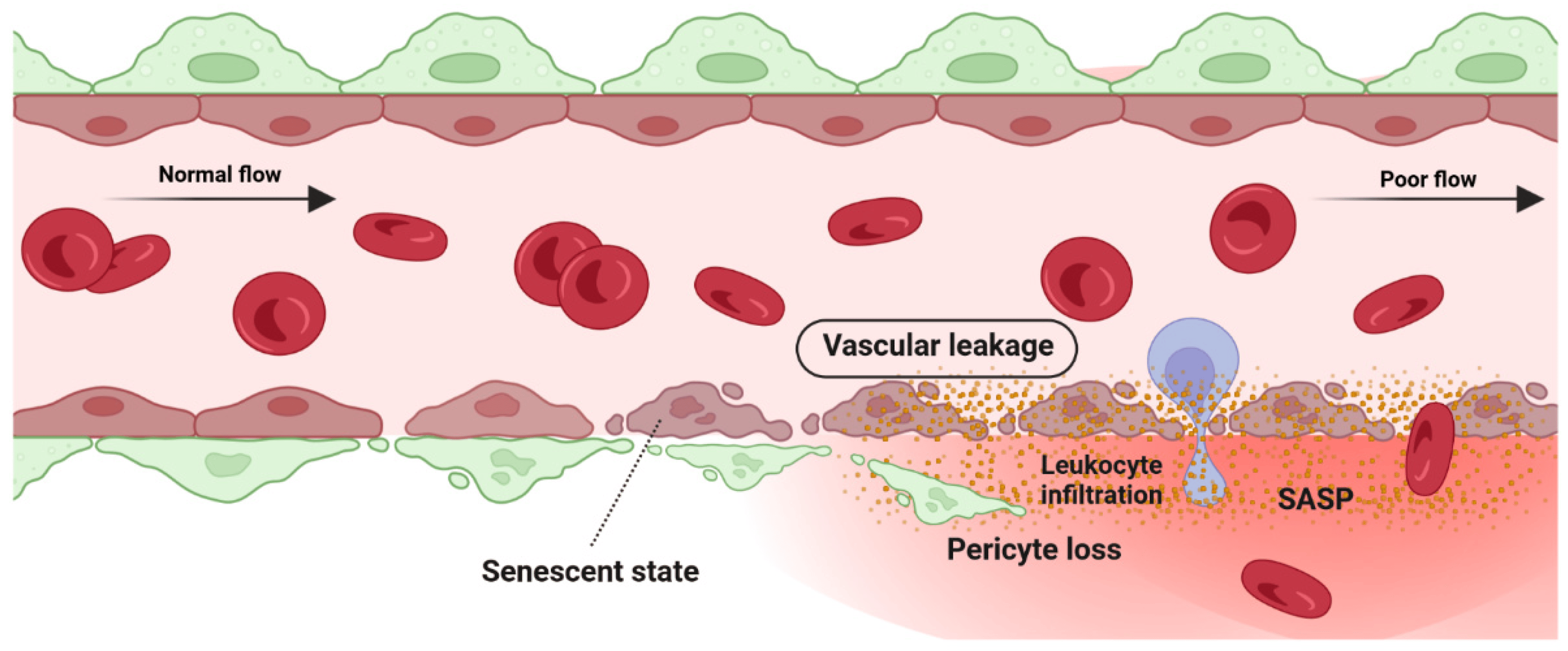
Figure 3. Detrimental effect: vascular degeneration. Senescence phenotype in pericyte and retinal endothelial cells is associated with pericyte detachment and vascular leakage.
2.2. Detrimental Effect: Pathological Angiogenesis
The hypoxic/oxidative nature of the ischemic retina triggers cellular senescence predominantly in the avascular zone to protect retinal cells from a low metabolic supply and hypoxia-associated cell death [53]. The senescence phenotype is not limited just to the avascular zone and there is a wealth of evidence showing the SASP’s engagement in propagating cellular senescence to the surrounding tissue in an autocrine and paracrine manner [64][65][66]. For instance, during the progression of retinopathy, secretion of the neuron-derived Semaphorin 3A (SEMA3A) by senescent retinal neuronal cells contributes to propagating paracrine senescence [53]. A recent study by Crespo-Garcia, Tsuruda, and Dejda et al. also demonstrated a cellular senescence burden at the peak of retinal neovascularization in a mouse model of oxygen-induced retinopathy (OIR) [67]. The high metabolic activity of the stimulated retinal SCs eventually promotes inflammation in the surrounding tissue microenvironment via the secretion of a pool of bioactive molecules (SASPs). These molecules include matrix-degrading proteases, growth factors, inflammatory chemokines, and cytokines. Further research by Oubaha et al. demonstrated that the ischemic retinal cells in retinopathies become prematurely senescent and exacerbate pathological angiogenesis by secreting a series of inflammatory cytokines [53]. Altogether, senescent retinal cells not only contribute to propagating the senescence phenotype in retinopathies, but also promote the destructive growth of new blood vessels from preexisting vessels (Figure 4).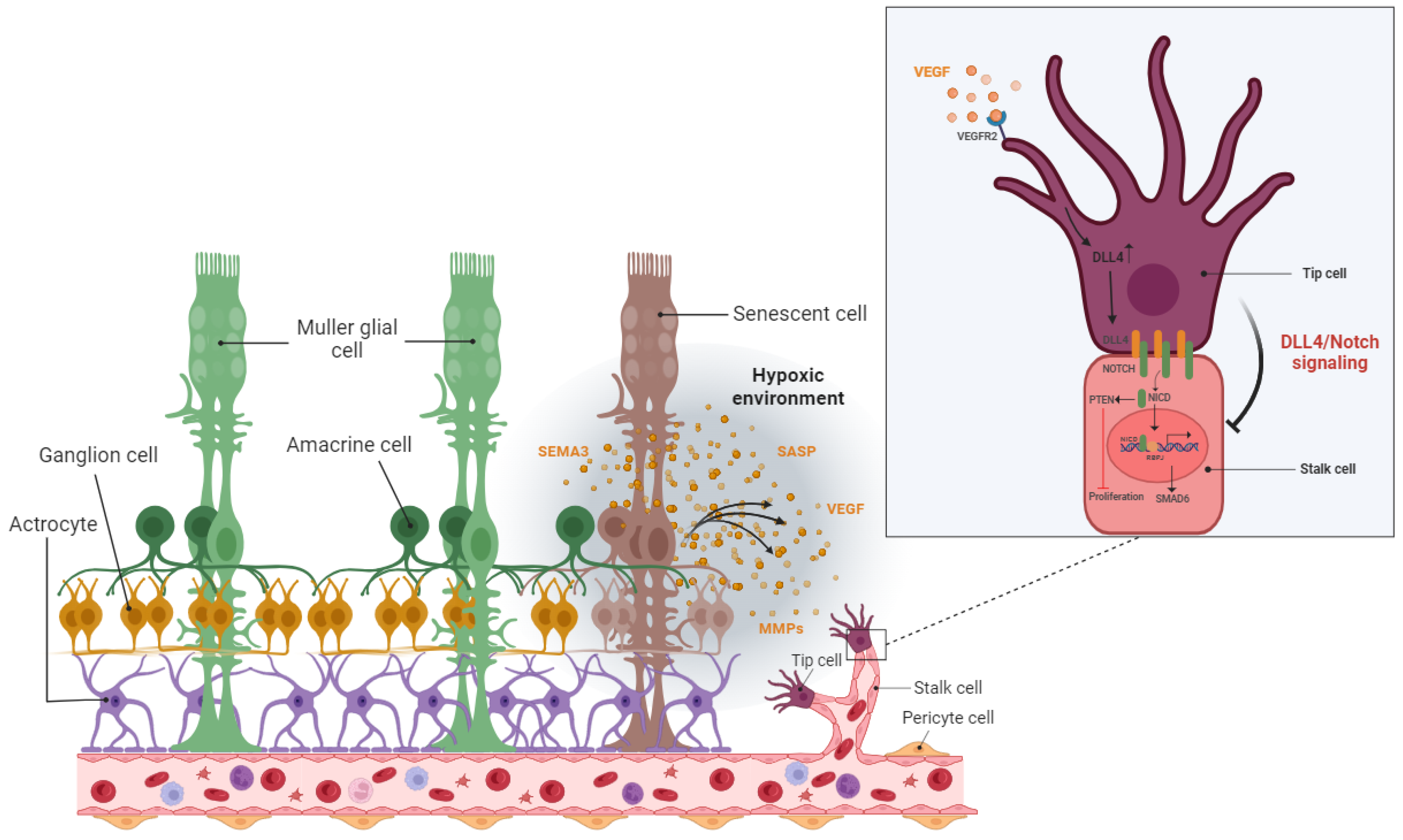
Figure 4. Detrimental effect: pathological angiogenesis. In the stressed retina, senescence neurons in the avascular area attract tip cells of the retinal blood vessel by releasing various proinflammatory and angiogenic factors and lead to dysregulated angiogenesis as a common feature of proliferative retinopathies.
2.3. Beneficial Effect: Vascular Remodeling
Vascular development and remodeling occur in the retina by executing two predefined physiological programs: (1) vascular growth (sprouting, proliferation, and branching and (2) vascular regression. In the fetal developing eyes, the programmed regression of vessels happens during the development of the hyaloid vasculature system. Failure in the regression of the hyaloid vessels causes severe intraocular hemorrhage, massive accumulation of vessels, and leads to persistent hyperplastic primary vitreous disease (PHPV) [68]. Physiological regression of blood vessels is not limited just to the retina but has also been discovered in the ductus arteriosus [69] and female reproductive system (during endometrial maturation) [70]. Vascular remodeling and regression were also detected in pathogenesis studies of several retinal disorders. Proliferative retinopathies (ROP, DR, and AMD) are associated with the pathological formation of a network of new blood vessels that are leaky, fragile, tortuous, and misdirected. Recent studies demonstrated the necessity of pruning pathological neovessels to prepare the retina for reparative vascular regeneration [53][71]. The spontaneous reparative vascular regression at the late stage of diseases usually attenuates the destructive costs and implications of the pathological neovessels [72]. This is supported by the fact that almost 90% of all affected ROP infants in the United States recover spontaneously without any therapeutic intervention [71][73][74]. Spontaneous regression of the pathological neovessels was also reported in DR individuals [75][76]. However, the cells and underlying mechanisms mediating vascular remodeling and regression in the retina have not been well defined. Vascular development and homeostasis is highly dependent upon a healthy endothelium [77]. The endothelium, a monolayer of vascular ECs, releases substances that are key modulators of matrix remodeling, vascular tone, inflammatory responses, and vascular cell proliferation [77]. Endothelial dysfunction is a common finding in all major cardiovascular diseases such as hypertension, atherosclerosis, and diabetes [78]. Recent studies also indicated that the metabolism of ECs regulates vascular growth (angiogenesis and vasculogenesis), and their central metabolism rewires during pathological vessel overgrowth [79]. Yoshikawa et al. (2015) even discovered that the regression rate of hyaloid vessels highly depends upon endothelial VEGFFR2 signaling [68]. It is therefore tempting to speculate that endothelial cells are possibly involved in the mechanisms mediating vascular remodeling and regression in the retina. Computational studies have now confirmed the enrichment of senescent markers in endothelial cells when preretinal neovascularization reaches its maximum level at P17 [53][67][71]. Senescence-associated secretory factors (e.g., TNF-α, IL-6, and MCP-1, and MMP) also play a leading role in matrix remodeling in vascular development and diseases [78]. SCs are resistant to apoptosis and generally are cleared by the immune system instead of necrotic or apoptotic mechanisms. The beneficial effect of the immune-mediated clearance of SCs in the progression of several developmental and physiological processes has been described recently (see Section 3). Impairment in the mechanisms of the SCs’ removal might result in cancer or aging-related disorders [80]. To date, a specific subpopulation of immune cells was shown to be recruited by SCs via SASP factors, this recruitment being essential to the elimination of SCs. For instance, in a model of liver fibrosis, senescent hepatic stellate cells promoted the resolution of fibrosis by attracting natural killer (NK) cells [81]. Antigen-specific CD4+ T cells orchestrate senescence surveillance of pre-malignant hepatocytes and prevent the development of murine hepatocellular carcinomas (HCCs) [82]. Abnormal accumulation of SCs in the postpartum uterus may impair subsequent pregnancy in mice [80]. Tissue-resident macrophages are another component of the immune system that is involved in the clearance of SCs. Egashira et al. (2017) provided evidence for the involvement of F4/80+ macrophages in the clearance of postpartum uterine senescence during the physiological process of uterine remodeling [80]. Similarly, in the retina, the transient presence of SCs is also beneficial, largely by recruiting immune cells for their elimination, but the prolonged presence of SCs can be deleterious. In ischemic retinopathies, the pathological neovascularization phase is followed by a phase of vascular remodeling and regression. However, the underlying mechanisms of the vascular remodeling and regression in the retina are ill-defined. Long-term exposure to the hyperglycemia, hypoxia, and inflammatory stress induces damage to the neurovascular cells including endothelial cells and pericytes [73]. The leaky, misdirected, and abnormal formed vessels in the neovascularization phase is not capable to fully supply the high oxygen and nutrient demands of the retina, leads to the generation of a tissue microenvironment with profound oxidative, cellular, and inflammatory stress [73]. A growing body of evidence supports the significance of the inflammation and pro-inflammatory mediators in driving proliferative retinopathies and pathological neovascularization [83][84][85][86]. Stressed retinal cells boost local inflammation and recruit immune cells to the ischemic retina [73]. Stressed endothelial cells, for example, enhance leukocyte adhesion and transmigration in microvessels via upregulating adhesion molecules (ICAM-1, VCAM-1, etc.) [87][88]. Recently, it has attracted much attention to explore the possible role of immune cells in the regression of pathological retinal neovessels (tufts). Recruited regulatory T cells to the ischemic retina repaired pathological neovascularization probably by altering activation state of microglia [83]. Inflammation and cellular stress in the retina stimulates activation and infiltration of mononuclear phagocytes (MPs) in the ischemic retina [73]. In this regard, studies in OIR model and in DR confirmed elevated number of MPs in the retina tissue [89][90]. Time dependent studies of the MPs distribution demonstrated that the number of M1-polarized MPs elevates during the phase of pathological neovascularization (starts at P12 and peaks at P17) while M2-polarized MPs population increases during the phase of vascular remodeling and regression (starts at P17 and peaks at P20) [91][92]. Recent studies suggested that MPs attenuate neovascularization and resolves existing pathological neovessels probably via killing (FasL-Fas interaction) and phagocyting stressed endothelial cells at pathological neovascular tufts [73][93][94][95][96]. Findings by Davies et al. also suggest that local secretion of MPs-mediating chemokines, such as MCP-1, promotes the resolution of pathologic neovessels by attracting retinal MPs to the neovascular tufts [97]. An interesting and recent study by Binet et al. led to important discoveries: (1) an enrichment of senescence and SASP markers in pericytes and ECs, (2) the arrival of neutrophils at the sites of pathological neovascular tufts when vascular regression begins (at P17), and (3) the detection of neutrophil extracellular traps (NETs) adjacent to the pathological neovessels [71]. Indeed, senescence-associated secretory phenotype of senescent ECs (e.g., IL-1β and CXCL1) attracts neutrophils to the pathological neovascular tufts and prompts the release of NETs [71]. Subsequently, NETs induce apoptosis in senescent ECs and promote regression of the pathological neovascular tufts in the ischemic retina [71] (Figure 5). Taken altogether, these findings demonstrated that inflammation and transient present of SCs at the late stage of proliferative retinopathies promotes regression of pathological neovessels and prepare ischemic retina for reparative vascular regeneration.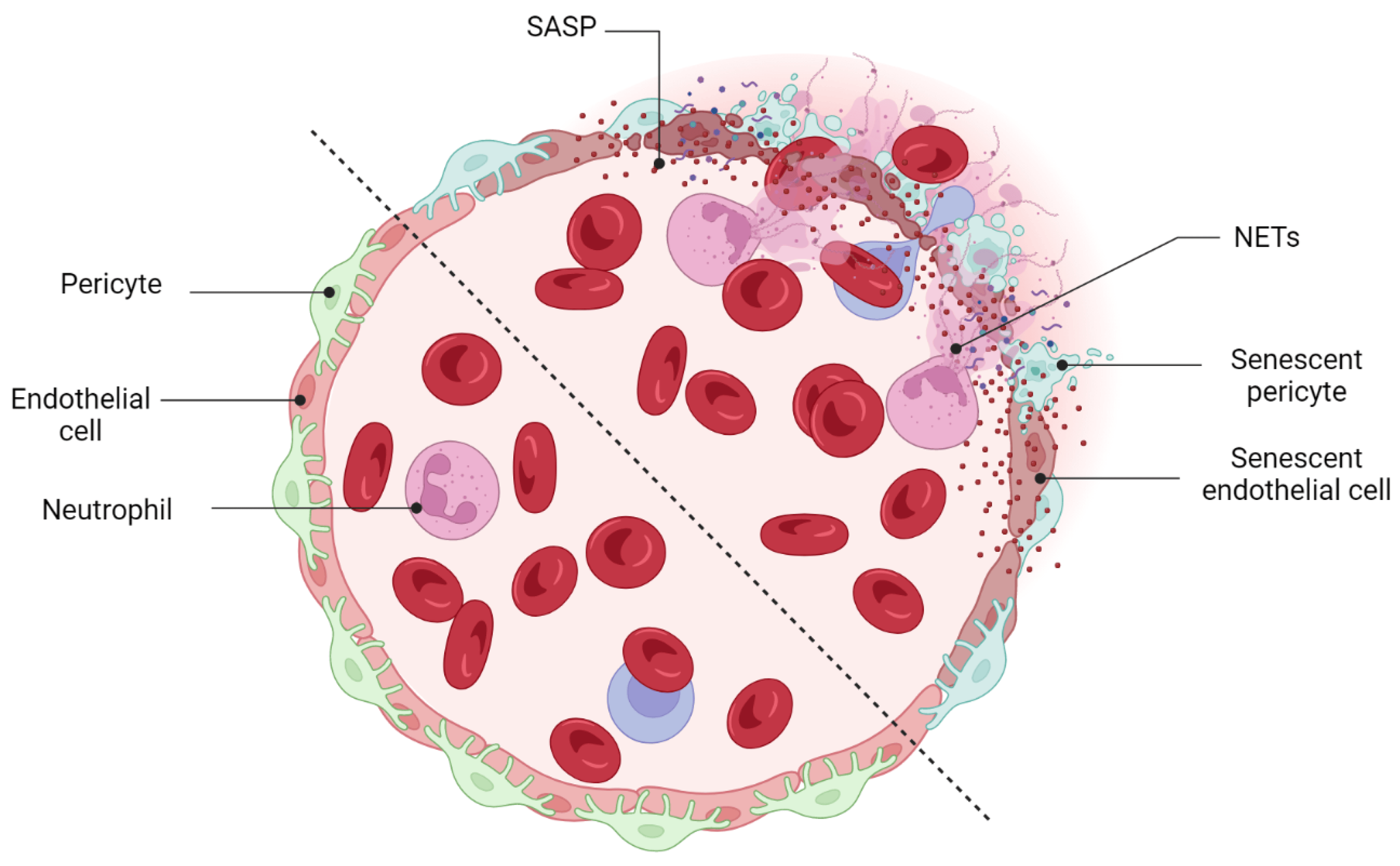
Figure 5. Beneficial effects of cellular senescence: vascular remodeling. Immune-mediated clearance of retinal SCs promotes the regression of pathological neovessels. Neutrophils recruit NETs (neutrophil extracellular traps) and prepare the retina for a reparative mechanism of regeneration by eliminating tuft-like neovessels.
References
- Kumari, R.; Jat, P. Mechanisms of Cellular Senescence: Cell Cycle Arrest and Senescence Associated Secretory Phenotype. Front. Cell Dev. Biol. 2021, 9, 645593.
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621.
- Vasileiou, P.V.S.; Evangelou, K.; Vlasis, K.; Fildisis, G.; Panayiotidis, M.I.; Chronopoulos, E.; Passias, P.G.; Kouloukoussa, M.; Gorgoulis, V.G.; Havaki, S. Mitochondrial Homeostasis and Cellular Senescence. Cells 2019, 8, 686.
- Matthews, H.K.; Bertoli, C.; de Bruin, R.A.M. Cell cycle control in cancer. Nat. Rev. Mol. Cell Biol. 2022, 23, 74–88.
- Zhao, M.L.; Rabiee, A.; Kovary, K.M.; Bahrami-Nejad, Z.; Taylor, B.; Teruel, M.N. Molecular Competition in G1 Controls When Cells Simultaneously Commit to Terminally Differentiate and Exit the Cell Cycle. Cell Rep. 2020, 31, 107769.
- Ruijtenberg, S.; van den Heuvel, S. Coordinating cell proliferation and differentiation: Antagonism between cell cycle regulators and cell type-specific gene expression. Cell Cycle 2016, 15, 196–212.
- Rumman, M.; Dhawan, J.; Kassem, M. Concise Review: Quiescence in Adult Stem Cells: Biological Significance and Relevance to Tissue Regeneration. Stem. Cells 2015, 33, 2903–2912.
- Maciel-Baron, L.A.; Moreno-Blas, D.; Morales-Rosales, S.L.; Gonzalez-Puertos, V.Y.; Lopez-Diazguerrero, N.E.; Torres, C.; Castro-Obregon, S.; Konigsberg, M. Cellular Senescence, Neurological Function, and Redox State. Antioxid Redox. Signal 2018, 28, 1704–1723.
- Cho, I.J.; Lui, P.P.; Obajdin, J.; Riccio, F.; Stroukov, W.; Willis, T.L.; Spagnoli, F.; Watt, F.M. Mechanisms, Hallmarks, and Implications of Stem Cell Quiescence. Stem Cell Rep. 2019, 12, 1190–1200.
- De Morree, A.; Rando, T.A. Regulation of adult stem cell quiescence and its functions in the maintenance of tissue integrity. Nat. Rev. Mol. Cell Biol. 2023, 24, 334–354.
- Salama, R.; Sadaie, M.; Hoare, M.; Narita, M. Cellular senescence and its effector programs. Genes Dev. 2014, 28, 99–114.
- Vergel, M.; Marin, J.J.; Estevez, P.; Carnero, A. Cellular senescence as a target in cancer control. J. Aging Res. 2010, 2011, 725365.
- Leikam, C.; Hufnagel, A.; Schartl, M.; Meierjohann, S. Oncogene activation in melanocytes links reactive oxygen to multinucleated phenotype and senescence. Oncogene 2008, 27, 7070–7082.
- Takahashi, A.; Ohtani, N.; Yamakoshi, K.; Iida, S.; Tahara, H.; Nakayama, K.; Nakayama, K.I.; Ide, T.; Saya, H.; Hara, E. Mitogenic signalling and the p16INK4a-Rb pathway cooperate to enforce irreversible cellular senescence. Nat. Cell Biol. 2006, 8, 1291–1297.
- Panopoulos, A.; Pacios-Bras, C.; Choi, J.; Yenjerla, M.; Sussman, M.A.; Fotedar, R.; Margolis, R.L. Failure of cell cleavage induces senescence in tetraploid primary cells. Mol. Biol. Cell 2014, 25, 3105–3118.
- Walen, K.H. Human diploid fibroblast cells in senescence; cycling through polyploidy to mitotic cells. Vitr. Cell Dev. Biol. Anim. 2006, 42, 216–224.
- Dikovskaya, D.; Cole, J.J.; Mason, S.M.; Nixon, C.; Karim, S.A.; McGarry, L.; Clark, W.; Hewitt, R.N.; Sammons, M.A.; Zhu, J.; et al. Mitotic Stress Is an Integral Part of the Oncogene-Induced Senescence Program that Promotes Multinucleation and Cell Cycle Arrest. Cell Rep. 2015, 12, 1483–1496.
- Rovira, M.; Sereda, R.; Pladevall-Morera, D.; Ramponi, V.; Marin, I.; Maus, M.; Madrigal-Matute, J.; Diaz, A.; Garcia, F.; Munoz, J.; et al. The lysosomal proteome of senescent cells contributes to the senescence secretome. Aging Cell 2022, 21, e13707.
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O.; et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 9363–9367.
- Severino, J.; Allen, R.G.; Balin, S.; Balin, A.; Cristofalo, V.J. Is beta-galactosidase staining a marker of senescence in vitro and in vivo? Exp. Cell Res. 2000, 257, 162–171.
- Terman, A.; Brunk, U.T. The aging myocardium: Roles of mitochondrial damage and lysosomal degradation. Heart Lung Circ. 2005, 14, 107–114.
- Brunk, U.T.; Terman, A. Lipofuscin: Mechanisms of age-related accumulation and influence on cell function. Free Radic Biol. Med. 2002, 33, 611–619.
- Sharpless, N.E.; Sherr, C.J. Forging a signature of in vivo senescence. Nat. Rev. Cancer 2015, 15, 397–408.
- Saraste, M. Oxidative phosphorylation at the fin de siecle. Science 1999, 283, 1488–1493.
- James, E.L.; Michalek, R.D.; Pitiyage, G.N.; de Castro, A.M.; Vignola, K.S.; Jones, J.; Mohney, R.P.; Karoly, E.D.; Prime, S.S.; Parkinson, E.K. Senescent human fibroblasts show increased glycolysis and redox homeostasis with extracellular metabolomes that overlap with those of irreparable DNA damage, aging, and disease. J. Proteome Res. 2015, 14, 1854–1871.
- Parkinson, E.K.; Adamski, J.; Zahn, G.; Gaumann, A.; Flores-Borja, F.; Ziegler, C.; Mycielska, M.E. Extracellular citrate and metabolic adaptations of cancer cells. Cancer Metastasis Rev. 2021, 40, 1073–1091.
- Zwerschke, W.; Mazurek, S.; Stockl, P.; Hutter, E.; Eigenbrodt, E.; Jansen-Durr, P. Metabolic analysis of senescent human fibroblasts reveals a role for AMP in cellular senescence. Biochem. J. 2003, 376, 403–411.
- Miwa, S.; Kashyap, S.; Chini, E.; von Zglinicki, T. Mitochondrial dysfunction in cell senescence and aging. J. Clin. Invest. 2022, 132, e158447.
- Passos, J.F.; Saretzki, G.; Ahmed, S.; Nelson, G.; Richter, T.; Peters, H.; Wappler, I.; Birket, M.J.; Harold, G.; Schaeuble, K.; et al. Mitochondrial dysfunction accounts for the stochastic heterogeneity in telomere-dependent senescence. PLoS Biol. 2007, 5, e110.
- Petersen, S.; Saretzki, G.; von Zglinicki, T. Preferential accumulation of single-stranded regions in telomeres of human fibroblasts. Exp. Cell Res. 1998, 239, 152–160.
- Manzella, N.; Santin, Y.; Maggiorani, D.; Martini, H.; Douin-Echinard, V.; Passos, J.F.; Lezoualc’h, F.; Binda, C.; Parini, A.; Mialet-Perez, J. Monoamine oxidase-A is a novel driver of stress-induced premature senescence through inhibition of parkin-mediated mitophagy. Aging Cell 2018, 17, e12811.
- Wiley, C.D.; Velarde, M.C.; Lecot, P.; Liu, S.; Sarnoski, E.A.; Freund, A.; Shirakawa, K.; Lim, H.W.; Davis, S.S.; Ramanathan, A.; et al. Mitochondrial Dysfunction Induces Senescence with a Distinct Secretory Phenotype. Cell Metab. 2016, 23, 303–314.
- Roger, L.; Tomas, F.; Gire, V. Mechanisms and Regulation of Cellular Senescence. Int. J. Mol. Sci. 2021, 22, 13173.
- Cormenier, J.; Martin, N.; Desle, J.; Salazar-Cardozo, C.; Pourtier, A.; Abbadie, C.; Pluquet, O. The ATF6alpha arm of the Unfolded Protein Response mediates replicative senescence in human fibroblasts through a COX2/prostaglandin E2 intracrine pathway. Mech. Ageing Dev. 2018, 170, 82–91.
- Ohno-Iwashita, Y.; Shimada, Y.; Hayashi, M.; Inomata, M. Plasma membrane microdomains in aging and disease. Geriatr. Gerontol. Int. 2010, 10 (Suppl 1), S41–S52.
- Narita, M.; Nunez, S.; Heard, E.; Narita, M.; Lin, A.W.; Hearn, S.A.; Spector, D.L.; Hannon, G.J.; Lowe, S.W. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell 2003, 113, 703–716.
- Di Micco, R.; Sulli, G.; Dobreva, M.; Liontos, M.; Botrugno, O.A.; Gargiulo, G.; dal Zuffo, R.; Matti, V.; d’Ario, G.; Montani, E.; et al. Interplay between oncogene-induced DNA damage response and heterochromatin in senescence and cancer. Nat. Cell Biol. 2011, 13, 292–302.
- d’Adda di Fagagna, F.; Reaper, P.M.; Clay-Farrace, L.; Fiegler, H.; Carr, P.; Von Zglinicki, T.; Saretzki, G.; Carter, N.P.; Jackson, S.P. A DNA damage checkpoint response in telomere-initiated senescence. Nature 2003, 426, 194–198.
- Reinhardt, H.C.; Schumacher, B. The p53 network: Cellular and systemic DNA damage responses in aging and cancer. Trends Genet. 2012, 28, 128–136.
- Smith, J.; Tho, L.M.; Xu, N.; Gillespie, D.A. The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Adv. Cancer Res. 2010, 108, 73–112.
- Weber, A.M.; Ryan, A.J. ATM and ATR as therapeutic targets in cancer. Pharmacol. Ther. 2015, 149, 124–138.
- Herbig, U.; Jobling, W.A.; Chen, B.P.; Chen, D.J.; Sedivy, J.M. Telomere shortening triggers senescence of human cells through a pathway involving ATM, p53, and p21(CIP1), but not p16(INK4a). Mol. Cell 2004, 14, 501–513.
- Serrano, M.; Hannon, G.J.; Beach, D. A new regulatory motif in cell-cycle control causing specific inhibition of cyclin D/CDK4. Nature 1993, 366, 704–707.
- Young, A.R.J.; Cassidy, L.D.; Narita, M. Autophagy and senescence, converging roles in pathophysiology as seen through mouse models. Adv. Cancer Res. 2021, 150, 113–145.
- Kwon, Y.; Kim, J.W.; Jeoung, J.A.; Kim, M.S.; Kang, C. Autophagy is pro-senescence when seen in close-up, but anti-senescence in long-shot. Mol. Cells 2017, 40, 607–612.
- Garcia-Prat, L.; Martinez-Vicente, M.; Perdiguero, E.; Ortet, L.; Rodriguez-Ubreva, J.; Rebollo, E.; Ruiz-Bonilla, V.; Gutarra, S.; Ballestar, E.; Serrano, A.L.; et al. Autophagy maintains stemness by preventing senescence. Nature 2016, 529, 37–42.
- Wen, X.; Klionsky, D.J. Autophagy is a key factor in maintaining the regenerative capacity of muscle stem cells by promoting quiescence and preventing senescence. Autophagy 2016, 12, 1158373.
- Kang, H.T.; Lee, K.B.; Kim, S.Y.; Choi, H.R.; Park, S.C. Autophagy impairment induces premature senescence in primary human fibroblasts. PLoS ONE 2011, 6, e23367.
- Young, A.R.; Narita, M.; Ferreira, M.; Kirschner, K.; Sadaie, M.; Darot, J.F.; Tavare, S.; Arakawa, S.; Shimizu, S.; Watt, F.M.; et al. Autophagy mediates the mitotic senescence transition. Genes Dev. 2009, 23, 798–803.
- Narita, M.; Young, A.R.; Arakawa, S.; Samarajiwa, S.A.; Nakashima, T.; Yoshida, S.; Hong, S.; Berry, L.S.; Reichelt, S.; Ferreira, M.; et al. Spatial coupling of mTOR and autophagy augments secretory phenotypes. Science 2011, 332, 966–970.
- Nam, H.Y.; Han, M.W.; Chang, H.W.; Kim, S.Y.; Kim, S.W. Prolonged autophagy by MTOR inhibitor leads radioresistant cancer cells into senescence. Autophagy 2013, 9, 1631–1632.
- Liu, H.; Prokosch, V. Energy Metabolism in the Inner Retina in Health and Glaucoma. Int. J. Mol. Sci. 2021, 22, 3689.
- Oubaha, M.; Miloudi, K.; Dejda, A.; Guber, V.; Mawambo, G.; Germain, M.A.; Bourdel, G.; Popovic, N.; Rezende, F.A.; Kaufman, R.J.; et al. Senescence-associated secretory phenotype contributes to pathological angiogenesis in retinopathy. Sci. Transl. Med. 2016, 8, 362ra144.
- Sapieha, P.; Mallette, F.A. Cellular Senescence in Postmitotic Cells: Beyond Growth Arrest. Trends Cell Biol. 2018, 28, 595–607.
- Brown, B.A.; Williams, H.; George, S.J. Evidence for the Involvement of Matrix-Degrading Metalloproteinases (MMPs) in Atherosclerosis. Prog. Mol. Biol. Transl. Sci. 2017, 147, 197–237.
- Bertelli, P.M.; Pedrini, E.; Hughes, D.; McDonnell, S.; Pathak, V.; Peixoto, E.; Guduric-Fuchs, J.; Stitt, A.W.; Medina, R.J. Long term high glucose exposure induces premature senescence in retinal endothelial cells. Front. Physiol. 2022, 13, 929118.
- Tian, X.L.; Li, Y. Endothelial cell senescence and age-related vascular diseases. J. Genet. Genom. 2014, 41, 485–495.
- Mylonas, A.; O’Loghlen, A. Cellular Senescence and Ageing: Mechanisms and Interventions. Front. Aging 2022, 3, 866718.
- Del Cuore, A.; Pacinella, G.; Riolo, R.; Tuttolomondo, A. The Role of Immunosenescence in Cerebral Small Vessel Disease: A Review. Int. J. Mol. Sci. 2022, 23, 7136.
- Cheung, T.M.; Ganatra, M.P.; Peters, E.B.; Truskey, G.A. Effect of cellular senescence on the albumin permeability of blood-derived endothelial cells. Am. J. Physiol. Heart Circ. Physiol. 2012, 303, H1374–H1383.
- Yamazaki, Y.; Baker, D.J.; Tachibana, M.; Liu, C.C.; van Deursen, J.M.; Brott, T.G.; Bu, G.; Kanekiyo, T. Vascular Cell Senescence Contributes to Blood-Brain Barrier Breakdown. Stroke 2016, 47, 1068–1077.
- Venkatesh, D.; Fredette, N.; Rostama, B.; Tang, Y.; Vary, C.P.; Liaw, L.; Urs, S. RhoA-mediated signaling in Notch-induced senescence-like growth arrest and endothelial barrier dysfunction. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 876–882.
- Krouwer, V.J.; Hekking, L.H.; Langelaar-Makkinje, M.; Regan-Klapisz, E.; Post, J.A. Endothelial cell senescence is associated with disrupted cell-cell junctions and increased monolayer permeability. Vasc. Cell 2012, 4, 12.
- Wajapeyee, N.; Serra, R.W.; Zhu, X.; Mahalingam, M.; Green, M.R. Oncogenic BRAF induces senescence and apoptosis through pathways mediated by the secreted protein IGFBP7. Cell 2008, 132, 363–374.
- Kuilman, T.; Michaloglou, C.; Vredeveld, L.C.; Douma, S.; van Doorn, R.; Desmet, C.J.; Aarden, L.A.; Mooi, W.J.; Peeper, D.S. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell 2008, 133, 1019–1031.
- Acosta, J.C.; O’Loghlen, A.; Banito, A.; Guijarro, M.V.; Augert, A.; Raguz, S.; Fumagalli, M.; Da Costa, M.; Brown, C.; Popov, N.; et al. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell 2008, 133, 1006–1018.
- Crespo-Garcia, S.; Tsuruda, P.R.; Dejda, A.; Ryan, R.D.; Fournier, F.; Chaney, S.Y.; Pilon, F.; Dogan, T.; Cagnone, G.; Patel, P.; et al. Pathological angiogenesis in retinopathy engages cellular senescence and is amenable to therapeutic elimination via BCL-xL inhibition. Cell Metab. 2021, 33, 818–832.e817.
- Yoshikawa, Y.; Yamada, T.; Tai-Nagara, I.; Okabe, K.; Kitagawa, Y.; Ema, M.; Kubota, Y. Developmental regression of hyaloid vasculature is triggered by neurons. J. Exp. Med. 2016, 213, 1175–1183.
- Hamrick, S.E.G.; Sallmon, H.; Rose, A.T.; Porras, D.; Shelton, E.L.; Reese, J.; Hansmann, G. Patent Ductus Arteriosus of the Preterm Infant. Pediatrics 2020, 146, e20201209.
- Girling, J.E.; Rogers, P.A. Regulation of endometrial vascular remodelling: Role of the vascular endothelial growth factor family and the angiopoietin-TIE signalling system. Reproduction 2009, 138, 883–893.
- Binet, F.; Cagnone, G.; Crespo-Garcia, S.; Hata, M.; Neault, M.; Dejda, A.; Wilson, A.M.; Buscarlet, M.; Mawambo, G.T.; Howard, J.P.; et al. Neutrophil extracellular traps target senescent vasculature for tissue remodeling in retinopathy. Science 2020, 369, eaay5356.
- Hartnett, M.E.; Penn, J.S. Mechanisms and management of retinopathy of prematurity. N. Engl. J. Med. 2012, 367, 2515–2526.
- Klotzsche-von Ameln, A.; Sprott, D. Harnessing retinal phagocytes to combat pathological neovascularization in ischemic retinopathies? Pflug. Arch. 2022, 474, 575–590.
- Ju, R.H.; Zhang, J.Q.; Ke, X.Y.; Lu, X.H.; Liang, L.F.; Wang, W.J. Spontaneous regression of retinopathy of prematurity: Incidence and predictive factors. Int. J. Ophthalmol. 2013, 6, 475–480.
- Han, J.R.; Ju, W.K.; Park, I.W. Spontaneous regression of neovascularization at the disc in diabetic retinopathy. Korean J. Ophthalmol. 2004, 18, 41–46.
- Bandello, F.; Gass, J.D.; Lattanzio, R.; Brancato, R. Spontaneous regression of neovascularization at the disk and elsewhere in diabetic retinopathy. Am. J. Ophthalmol. 1996, 122, 494–501.
- Gao, Y.; Galis, Z.S. Exploring the Role of Endothelial Cell Resilience in Cardiovascular Health and Disease. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 179–185.
- Melo, L.G.; Gnecchi, M.; Ward, C.A.; Dzau, V.J. Vascular Remodeling in Health and Disease. J. Cardiovasc. Med. 2007, 1541–1565.
- Eelen, G.; de Zeeuw, P.; Treps, L.; Harjes, U.; Wong, B.W.; Carmeliet, P. Endothelial Cell Metabolism. Physiol. Rev. 2018, 98, 3–58.
- Egashira, M.; Hirota, Y.; Shimizu-Hirota, R.; Saito-Fujita, T.; Haraguchi, H.; Matsumoto, L.; Matsuo, M.; Hiraoka, T.; Tanaka, T.; Akaeda, S.; et al. F4/80+ Macrophages Contribute to Clearance of Senescent Cells in the Mouse Postpartum Uterus. Endocrinology 2017, 158, 2344–2353.
- Nacher, V.; Carretero, A.; Navarro, M.; Armengol, C.; Llombart, C.; Rodriguez, A.; Herrero-Fresneda, I.; Ayuso, E.; Ruberte, J. The quail mesonephros: A new model for renal senescence? J. Vasc. Res. 2006, 43, 581–586.
- Kang, T.W.; Yevsa, T.; Woller, N.; Hoenicke, L.; Wuestefeld, T.; Dauch, D.; Hohmeyer, A.; Gereke, M.; Rudalska, R.; Potapova, A.; et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature 2011, 479, 547–551.
- Deliyanti, D.; Talia, D.M.; Zhu, T.; Maxwell, M.J.; Agrotis, A.; Jerome, J.R.; Hargreaves, E.M.; Gerondakis, S.; Hibbs, M.L.; Mackay, F.; et al. Foxp3(+) Tregs are recruited to the retina to repair pathological angiogenesis. Nat. Commun. 2017, 8, 748.
- Simo, R.; Hernandez, C. Novel approaches for treating diabetic retinopathy based on recent pathogenic evidence. Prog. Retin. Eye Res. 2015, 48, 160–180.
- Agarwal, A.; Rhoades, W.R.; Hanout, M.; Soliman, M.K.; Sarwar, S.; Sadiq, M.A.; Sepah, Y.J.; Do, D.V.; Nguyen, Q.D. Management of neovascular age-related macular degeneration: Current state-of-the-art care for optimizing visual outcomes and therapies in development. Clin. Ophthalmol. 2015, 9, 1001–1015.
- Sapieha, P.; Joyal, J.S.; Rivera, J.C.; Kermorvant-Duchemin, E.; Sennlaub, F.; Hardy, P.; Lachapelle, P.; Chemtob, S. Retinopathy of prematurity: Understanding ischemic retinal vasculopathies at an extreme of life. J. Clin. Invest. 2010, 120, 3022–3032.
- Altmann, C.; Schmidt, M.H.H. The Role of Microglia in Diabetic Retinopathy: Inflammation, Microvasculature Defects and Neurodegeneration. Int. J. Mol. Sci. 2018, 19, 110.
- Semeraro, F.; Cancarini, A.; dell’Omo, R.; Rezzola, S.; Romano, M.R.; Costagliola, C. Diabetic Retinopathy: Vascular and Inflammatory Disease. J. Diabetes Res. 2015, 2015, 582060.
- Arroba, A.I.; Alcalde-Estevez, E.; Garcia-Ramirez, M.; Cazzoni, D.; de la Villa, P.; Sanchez-Fernandez, E.M.; Mellet, C.O.; Garcia Fernandez, J.M.; Hernandez, C.; Simo, R.; et al. Modulation of microglia polarization dynamics during diabetic retinopathy in db/db mice. Biochim. Biophys. Acta 2016, 1862, 1663–1674.
- Zeng, H.Y.; Green, W.R.; Tso, M.O. Microglial activation in human diabetic retinopathy. Arch. Ophthalmol. 2008, 126, 227–232.
- Li, J.; Yu, S.; Lu, X.; Cui, K.; Tang, X.; Xu, Y.; Liang, X. The phase changes of M1/M2 phenotype of microglia/macrophage following oxygen-induced retinopathy in mice. Inflamm. Res. 2021, 70, 183–192.
- Zhou, Y.; Yoshida, S.; Nakao, S.; Yoshimura, T.; Kobayashi, Y.; Nakama, T.; Kubo, Y.; Miyawaki, K.; Yamaguchi, M.; Ishikawa, K.; et al. M2 Macrophages Enhance Pathological Neovascularization in the Mouse Model of Oxygen-Induced Retinopathy. Investig. Ophthalmol. Vis. Sci. 2015, 56, 4767–4777.
- Korovina, I.; Neuwirth, A.; Sprott, D.; Troullinaki, M.; Poitz, D.M.; Deussen, A.; Klotzsche-von Ameln, A. Myeloid SOCS3 Deficiency Regulates Angiogenesis via Enhanced Apoptotic Endothelial Cell Engulfment. J. Innate Immun. 2020, 12, 248–256.
- Korovina, I.; Neuwirth, A.; Sprott, D.; Weber, S.; Sardar Pasha, S.P.B.; Gercken, B.; Breier, G.; El-Armouche, A.; Deussen, A.; Karl, M.O.; et al. Hematopoietic hypoxia-inducible factor 2alpha deficiency ameliorates pathological retinal neovascularization via modulation of endothelial cell apoptosis. FASEB J. 2019, 33, 1758–1770.
- He, P. Leucocyte/endothelium interactions and microvessel permeability: Coupled or uncoupled? Cardiovasc. Res. 2010, 87, 281–290.
- Joussen, A.M.; Poulaki, V.; Mitsiades, N.; Cai, W.Y.; Suzuma, I.; Pak, J.; Ju, S.T.; Rook, S.L.; Esser, P.; Mitsiades, C.S.; et al. Suppression of Fas-FasL-induced endothelial cell apoptosis prevents diabetic blood-retinal barrier breakdown in a model of streptozotocin-induced diabetes. FASEB J. 2003, 17, 76–78.
- Davies, M.H.; Stempel, A.J.; Powers, M.R. MCP-1 deficiency delays regression of pathologic retinal neovascularization in a model of ischemic retinopathy. Invest. Ophthalmol. Vis. Sci. 2008, 49, 4195–4202.
- Wang, L.; Zhou, X.; Yin, Y.; Mai, Y.; Wang, D.; Zhang, X. Hyperglycemia Induces Neutrophil Extracellular Traps Formation Through an NADPH Oxidase-Dependent Pathway in Diabetic Retinopathy. Front. Immunol. 2018, 9, 3076.
- Wong, S.L.; Demers, M.; Martinod, K.; Gallant, M.; Wang, Y.; Goldfine, A.B.; Kahn, C.R.; Wagner, D.D. Diabetes primes neutrophils to undergo NETosis, which impairs wound healing. Nat. Med. 2015, 21, 815–819.
- Menegazzo, L.; Ciciliot, S.; Poncina, N.; Mazzucato, M.; Persano, M.; Bonora, B.; Albiero, M.; Vigili de Kreutzenberg, S.; Avogaro, A.; Fadini, G.P. NETosis is induced by high glucose and associated with type 2 diabetes. Acta Diabetol. 2015, 52, 497–503.
- Park, J.H.; Kim, J.E.; Gu, J.Y.; Yoo, H.J.; Park, S.H.; Kim, Y.I.; Nam-Goong, I.S.; Kim, E.S.; Kim, H.K. Evaluation of Circulating Markers of Neutrophil Extracellular Trap (NET) Formation as Risk Factors for Diabetic Retinopathy in a Case-Control Association Study. Exp. Clin. Endocrinol. Diabetes 2016, 124, 557–561.
More