Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Lindsay Dong and Version 1 by Himadri Biswas.
The DNA damage response (DDR) is recognized as having an important role in cancer growth and treatment. ATR (ataxia telangiectasia mutated and Rad3-related) kinase, a major regulator of DDR, has shown significant therapeutic potential in cancer treatment. ATR inhibitors have shown anti-tumor effectiveness, not just as monotherapies but also in enhancing the effects of standard chemotherapy, radiation, and immunotherapy.
- ATR
- DNA damage responses
- DNA damage checkpoint signaling
- embryogenesis
- tumorigenesis
- apoptosis
- cancer therapeutics
1. Introduction
Most malignancies have genomic instability, which can cause oncogenesis. In response to DNA damage, cells have evolved several methods to protect their genome. These include the checkpoint mechanism, which protects the genome by limiting cell cycle progression in the event of DNA damage. ATR (ATM and Rad3-related), a member of the PIKK (phosphatidyl inositol 3′ kinase-related kinases) protein family, is a crucial protein in checkpoint responses. ATR is activated by both DNA-damaging agents and halted replication forks. The formation of DNA damage-induced elongation replication protein A (RPA)-coated single-stranded DNA (RPA-ssDNA) during replication stressors and during DNA repair is a common theme that leads to ATR activation [1,2][1][2]. ATR interacts with a nuclear partner, ATRIP, to form the ATR–ATRIP complex, which is recruited to the sites of DNA damage by RPA-ssDNA [3,4][3][4]. The recruitment of the ATR–ATRIP complex (ATR–ATRIP) to RPA-ssDNA, on the other hand, is insufficient for ATR activation. ATR is autophosphorylated at its T1989 residue [5] after being recruited to DNA damage sites. TopBP1 (topoisomerase 2 binding protein 1) binds at this phosphorylated residue and in turn increases ATR kinase activity [6]. At dsDNA-ssDNA junctions, TopBP1 appears to interact with the Rad9–Rad1–Hus1 (9-1-1) complex [7,8,9][7][8][9]. TopBP1 then directly stimulates the ATR–ATRIP kinase [7,8,10][7][8][10]. The RPA binding protein, Ewing’s tumor-associated antigen 1 (ETAA1), interacts with RPA and functions at stalled replication forks [11,12][11][12]. ETAA1, like TopBP1, directly activates ATR–ATRIP [11,12][11][12]. In the case of severe damage, activated ATR also stimulates the activities of several key downstream proteins, e.g., p53 and other checkpoint kinases such as Chk1, resulting in an S phase cell cycle interruption to repair DNA damage or apoptosis [2,5,7,8,12,13,14,15,16,17][2][5][7][8][12][13][14][15][16][17] (Figure 1). The substrates and effectors of ATR and Chk1 have been identified to be a growing collection of DNA replication, repair, or cell cycle proteins. In particular, Chk1 phosphorylation of Cdc25 phosphatases is relevant to DNA damage inducing cancer cell cycle arrest [18]. To correct DNA replication in cells exposed to replication stress, the phosphorylation by ATR of WRN, SMARCAL1, and FANCI is essential [19,20,21][19][20][21]. ATR is also involved in the regulation of numerous DNA repair mechanisms, including homologous recombination, inter-strand crosslink repair, and nucleotide excision repair [22,23,24,25,26][22][23][24][25][26].
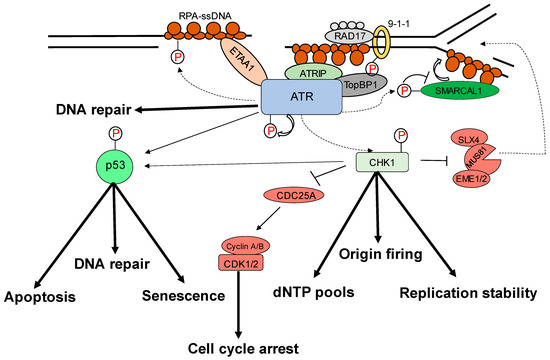
Figure 1. Replication stress-induced ATR–CHK1 activation. ATR is activated by RPA-coated single-stranded DNA (ssDNA) that forms at a stalled replication fork or resected DNA double-strand break (DSB), particularly at the ssDNA/dsDNA confluence. ATR-interacting protein (ATRIP) recruitment results in ATR and RPA-ssDNA complex recognition. It then integrates Rad9–Rad1–hus1 (9-1-1) and DNA topoisomerase 2-binding protein 1 (TOPBP1), activating ATR. ATR phosphorylates checkpoint kinase 1 (CHK1) via the adaptor protein claspin. CHK1 activation can help to prevent genomic instability. The processes promote or prevent the initiation of DNA replication (origin firing), ensure a sufficient supply of deoxynucleotides (dNTPs), stabilize the replication fork, and repair DNA. The signal is sent by transducer proteins to effector proteins such as p53, which is phosphorylated by ATR and CHK1. Cell-cycle arrest, DNA repair, apoptosis, and senescence are all mediated by p53.
2. Role of ATR–Chk1 Pathway in Oncogenesis
In mouse models, homozygous ATR deletion is embryonically lethal, and in mouse embryonic fibroblasts that are acutely genetically inactivated by ATR, one or two rounds of DNA replication are required to leave the cell cycle permanently [4,28][4][27]. In contrast, a mouse model expressing kinase-dead ATR (ATR+/KD) but not ATR loss (ATR−/−) exhibited ssDNA-dependent defects at the non-homologous region of X–Y chromosomes during male meiosis, resulting in sterility, and at telomeres, rDNA, and fragile sites during mitosis, resulting in lymphocytopenia [29][28]. Prompt ATR exchange at DNA damage sites requires ATR kinase activity, resulting in enhancement of Chk1 phosphorylation. ATR-KD traps a fraction of ATR and RPA to chromatin, where ATM/DNA-PKcs hyperphosphorylate RPA and prevent subsequent repair [29][28]. Because of the replication stress resulting from spontaneously defective DNA, and particularly hard to replicate areas in the genome such as fragile regions, an ATR pathway is essential. Furthermore, genetic inactivation of ATR in adult mice causes premature aging, abnormalities in tissue homeostasis, and progenitor cell depletion in high proliferative regions [30,31][29][30].
ATR, on the other hand, is more critical in many tumor cells than it is in normal ones. First, oncoproteins such as the Ras isoforms, Myc, and Cyclin E induce dysregulated signaling that disrupts normal cell cycle regulation and causes replication stress [34][31]. The ATR pathway plays an important role in the survival of these tumor cells, and several studies have shown that blocking this pathway has a preferentially lethal effect on cells with high levels of oncogene-induced replication stress [33,35,36,37,38,39,40,41][32][33][34][35][36][37][38][39]. Second, deficiency in ATM makes the cells more sensitive to ATR inhibition in cell culture and in animal models [42[40][41][42],43,44], a finding that has led to clinical trials of an ATR inhibitor in tumors characterized by ATM deficiency or ATM mutations. Third, in cell line models, loss of specific DNA repair proteins (e.g., XRCC1, ERCC1) causes tumor cell lines more sensitive to ATR inhibition [45,46,47,48][43][44][45][46]; however, these findings are yet to be implemented in animal models. Fourth, hypoxic cells, which are typically resistant to chemo- and radiation therapy [49][47], are sensitive to ATR inhibition [50,51[48][49][50],52], likely because hypoxia causes replication stress [53][51]. Fifth, tumor cells that rely on the alternative lengthening of telomeres (ALT) pathway are also more sensitive to ATR disruption due to ATR’s role in the homologous recombination reactions that maintain these telomeres [54][52]. Thus, taken together, these observations indicate that multiple events that drive tumorigenesis may create a synthetic lethality for ATR inhibition such as the finding that PARP inhibition is synthetically lethal in BRCA1/2-deficient tumors [55][53].
3. Cytoplasmic Functions of ATR
Apart from ATR nuclear function regulated through the kinase domain, the cytoplasmic ATR was discovered to serve a significant antiapoptotic effect directly at the mitochondria, independent of nuclear ATR and kinase activity [63][54]. The nuclear kinase and cytoplasmic antiapoptotic functions of ATR are carried out independently by two prolyl isomeric forms of ATR, namely, trans- and cis-ATRS428P429 [63][54]. Both of these variants rely on a single-peptide bond orientation in ATR via a prolyl isomerization motif at S428P429 in humans. ATRIP is consistently missing in the cytoplasm, except during its production. In contrast to nuclear ATR, which is constantly in the trans form in a complex with ATRIP, cytoplasmic ATR, which lacks ATRIP, is mostly in the cis isomeric form after DNA damage [63][54]. Pin1 and PP2A balance the cis and trans cytoplasmic versions of ATR, with the former facilitating the conversion of cis- to trans-ATR by detecting the phosphorylated Serine 428–Proline 429 motif (pS428-P429) in ATR’s N-terminus [63,64][54][55]. Although Pin 1 activity promotes trans-ATR synthesis, DAPKI kinase inactivates it in response to DNA damage. In addition, DNA damage activates PP2A, which dephosphorylates ATR (pS428-P429), not only disabling the S428 phosphorylation-dependent Pin1 activity on ATR but also leading to cis-ATR formation ATR [64[55][56],65], as ATR is naturally stable in the cis-isomeric form of ATR [65][56]. Unlike the trans-ATR isoform, the cis-ATR includes an exposed BH3-like domain, which allows it to connect to the proapoptotic tBid protein at the mitochondria [63,64,65,66,67][54][55][56][57][58] (Figure 2). This binding prevents tBid from triggering the Bax–Bak polymerization pathway, which is necessary for the intrinsic apoptotic pathway. From here on, cis-ATR performs an antiapoptotic function, allowing the cell to survive long enough to repair its damaged DNA [64][55]. It is of importance to investigate whether cis-ATR might be used as a potential target for developing effective novel anticancer drugs.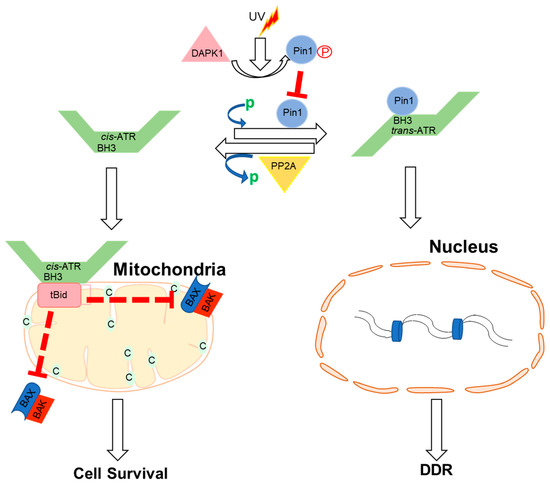
Figure 2. Proposed mechanisms by which ATR plays a direct anti-apoptotic function at the mitochondria. UV irradiation inhibits Pin1’s isomerization of ATR in the cytoplasm. Cis-ATR (ATR-H) then accumulates at the outer mitochondrial membrane, where it binds to and sequesters t-Bid. Because Bax and Bak cannot polymerize in the absence of tBid, cis-ATR suppresses cytochrome c release and apoptosis. In the nucleus, trans-ATR is the dominant isomer, interacting with ATRIP, RPA, and chromatin in the DNA damage repair (DDR) response. Protein phosphatases (PP2A) can dephosphorylate the Pin1 recognition motif and induce cis-ATR synthesis.
4. ATR Regulation of Nucleus Mechanics
Nuclear mechanosensing and mechanical features of the nucleus influence genome integrity, nuclear architecture, gene expression, cell migration, and differentiation [68,69][59][60]. ATR is a transcription factor that responds to mechanical stress at the nuclear envelope and mediates the envelope-associated repair of aberrant topological DNA states. Alteration of nuclear flexibility and YAP delocalization is observed in ATR-defective cells. ATR-defective nuclei collapse when subjected to mechanical stress or interstitial migration, accumulating nuclear envelope ruptures and perinuclear cGAS, indicating loss of nuclear envelope integrity and an abnormal perinuclear chromatin state. Furthermore, during development and in the metastatic spread of circulating tumor cells, ATR-defective cells have a defect in neuronal migration. Furthermore, the mechanical coupling of the cytoskeleton to the nuclear envelope and the accompanying regulation of the envelope–chromosome association is ensured by ATR [70][61].
5. ATR Regulation of Autophagy
DNA damage has been linked to autophagy via ATR/Chk1/RhoB-mediated lysosomal recruitment of the TSC complex and subsequent mTORC1 suppression. UV light (UV) or the alkylating chemical methyl methane sulphonate (MMS) damage DNA, causing Chk1 to phosphorylate the small GTPase RhoB. Phosphorylation of RhoB boosts its interaction with TSC2 and sumoylation by PIAS1, both of which are essential for the RhoB/TSC complex to translocate to lysosomes. mTORC1 is thereby blocked, and autophagy is induced. RhoB knockout drastically reduces TSC complex lysosomal translocation and DNA damage-induced autophagy, both of which are dependent on ATR–Chk1-mediated RhoB phosphorylation and sumoylation [71][62]. By acting on the transcriptional factor GATA4, ATR has also been associated with transcription regulation [72][63], which is susceptible to degradation by the autophagy-related factor p62 (also known as sequestosome 1 (SQSTM1)). In fibroblasts, ATR has been reported to have a negative effect on p62 [73][64]. In addition, a reduction in GATA4 levels and changes in gene expression associated with senescence and inflammation are observed when cells are treated with an ATR inhibitor [73][64].
6. The cis-ATR Anti-Apoptotic Role Drives Oncogenesis in Dividing Cells
Cancer is characterized by deregulated cell proliferation, which develops when there is an imbalance in the normal cell cycle regulation to govern the pace and integrity of cell division and growth. Furthermore, because cis-ATR has antiapoptotic properties, scholars surmise that it may have an oncogenic role, whereas Pin1 may have tumor-suppressive properties in relation to ATR’s anti-apoptotic activity at the mitochondria. If cis-ATR is the dominant cytoplasmic form, it may inhibit mitochondrial apoptosis, allowing injured cells to survive and mutate even when DNA damage repair is insufficient, and the aberrant cells are intended to die by apoptosis. This evasion of apoptosis is a key feature of cancer cells, allowing them to accumulate the mutations that define genomic instability and, eventually, carcinogenesis. However, if Pin1 activity is elevated and trans-ATR is the dominant form of ATR in the cytoplasm, programmed death will occur in cells that are too badly damaged for effective DNA repair before mutations can be transmitted. Thus, lowering cytosolic cis-ATR prevents the accumulation of cells with DNA damage, which could be passed on to daughter cells and induce carcinogenesis. The current knowledge is based mostly on data showing that Pin1 is overexpressed/activated in most malignancies and cancer stem cells, with correspondingly poor prognoses [72,74,75,76,77,78,79,80][63][65][66][67][68][69][70][71]. Pin1 also promotes the expression of multiple oncogenes while suppressing the expression of several tumor suppressor genes [81][72]. Pin1 overexpression or activation can be blocked genetically or chemically with juglone [82][73], all-trans retinoic acid [34][31] (ATRA), or KPT-6566 [83][74], and Pin1 inhibitors have been shown to reduce malignancies when tested [84,85,86,87,88,89,90][75][76][77][78][79][80][81]. However, chemically suppressing Pin1 presents numerous complications, particularly with retinoids (e.g., ATRA), the most used clinical inhibitor. Low medication bioavailability, clinical relapse, and retinoid resistance are examples of these [91,92,93,94,95][82][83][84][85][86]. Following UV-induced DNA damage, the phosphatase PP2A dephosphorylates ATR at Ser428. Following UV irradiation, PP2A was shown to be involved in the decrease of pATR (S428) in the cytoplasm [64][55]. Because Pin1 function needs the Ser428–Pro429 recognition site in ATR to be phosphorylated, PP2A inhibits Pin1 activity by depleting cytoplasmic pATR at S428. Another layer of regulatory complexity to the DDR process is associated with this PP2A monitoring for ATR phosphorylation at the Pin1 motif recognition. In the event of DNA damage, PP2A interacts with ATR to dephosphorylate its Pin 1 recognition motif to prevent further isomerization of cis to trans ATR in the cytoplasm, resulting in accumulation of cis ATR-H in the cytoplasm. It was found that the accumulation of cis ATRH in the cytoplasm and mitochondria, following DNA damage, is associated with PP2A activity. Mitochondrial translocation ATR-H is an antiapoptotic protein that interacts with tBid to inhibit apoptosis activation and to reduce apoptotic cell death caused by DNA damage [64][55].7. ATR as Therapeutic Target
7.1. Chemotherapeutic Effect by ATR–CHK1 Inhibition
The ability of ATR to limit oncogenesis in its early stages, by acting as a barrier to the proliferation of aberrant cells, occurs principally through p53 activation. The activation may lead to DNA repair and checkpoint arrest [102][87]. The fundamental theory is that ATR/CHK1 inhibitors, particularly in p53-deficient cells, increase tumor cell death by cytotoxic drugs or radiation by interrupting cell cycle checkpoints [103,104][88][89]. UCN-01 (7-hydroxystaurosporine) is the first CHK1 inhibitor with broad-spectrum activity against the protein kinase C family. In addition, UCN-01 lacks selectivity and has a long half-life, limiting its potential applications, since it binds to alpha acidic glycoprotein, which causes hyperglycemia, and has a long half-life [105,106][90][91]. XL844, an ATP-competitive inhibitor of CHK1, CHK2, VEGFR-2, and VEGFR-3, is quite effective. XL844 is designed to prevent CDC25A degradation, bypass S-Phase checkpoints, and increase DNA damage when used with gemcitabine. In the case of xenografts and in vitro, XL844 increases gemcitabine activity. The clinical study of XL844NCT00479175 and NCT00234481 was called off for reasons that are not yet known [107,108][92][93].7.2. ATR Kinase Inhibitors
ATR belongs to the PI3K-related kinase (PIKK) family of enzymes, along with ATM, mTOR, DNA-PKcs, SMG1, and PI3K (Phosphatidyl Inositol 3 Kinase). Caffeine was the first ATR inhibitor to be identified that disrupted DNA damage induced cell cycle arrest and sensitized cells with DNA damage [117,128][94][95]. However, since the concentrations that prevent ATR and ATM are toxic, this medicine cannot be used in clinical trials, as it is also preventing ATM and PI3K family members. The first potent and selective inhibitor, VE821, was reported by Vertex Pharmaceuticals in 2011. In comparison to ATM, PI3K, DNA-PK, and mTOR [129][96], VE-821 has >100-fold greater selectivity for ATR, and its analog VE-822 (VX-970) has been further enhanced with higher solubility, potency, selectivity, and pharmacodynamic characteristics. Preclinical studies showed that these agents potently sensitize several cancer cells lines to cisplatin, ionizing radiation, gemcitabine, PARP inhibitors, and topoisomerase I poisons etoposide and oxaliplatin in vitro [130,131,132,133,134,135][97][98][99][100][101][102]. Very selective, potent ATR inhibitors are also in development by AstraZeneca. In 2103, AZ20 was first reported [43][41]. The analog of AZ20, AZD6738, was described in 2013 as an orally accessible drug with improved solubility and pharmacodynamic qualities [136][103]. Compared to other types of PIKK, AZD6738’s selectivity is excellent for ATR [137][104].7.3. ATR Kinase Inhibitors in Clinical Trials
In 2009, the first report on ATR-selective small-molecule inhibitors was published [118][105]. Schisandrin B, a naturally occurring dibenzo cyclooctadiene lignan found in the medicinal herb Schisandra chinensis, was found to be a selective inhibitor of ATR [118][105]. Schisandrin B inhibited UV-induced intra-S-phase and G2/M cell cycle checkpoints, and the cytotoxicity in human lung cancer cells was increased upon UV radiation. The inhibitory potency against ATR, on the other hand, was modest and required the use of large drug concentrations (30 M for cellular tests). A more potent ATR inhibitor, NU6027, was reported in 2011 and was demonstrated to sensitize several breast and ovarian cancer cell lines to IR (insulin resistance) and several chemotherapeutic agents. However, this drug was originally created as a CDK2 inhibitor and is not ATR selective. Toledo et al. also published the findings of a cell-based chemical library screening technique for the identification of effective ATR inhibitors in 2011 [34][31]. One of the compounds identified to possess significant inhibitory activity against ATR kinase was NVP-BEZ235, a drug originally introduced as a highly potent dual inhibitor of PI3K and MTOR with considerable in vivo anti-tumor activity [140][106]. NVP-BEZ235 has been demonstrated to be markedly radiosensitive to Ras-overexpressing tumors [141][107]. Vertex Pharmaceuticals discovered the first class of effective and selective ATR kinase inhibitors during a high-throughput screening strategy [133][100]. VE-821 was found to be a powerful inhibitor of ATP-like effects on ATR, but it had little to no interaction with other PIKKs such as ATM, DNA, and MTOR [44][42]. In the colorectal cancer cell line HCT116, VE-821 reduced phosphorylation of the ATR downstream target CHK1 at Ser345 and showed excellent constructive collaboration with genotoxic drugs from several classes. Chemosensitization was particularly evident with DNA cross-linking agents such as cisplatin, and it was further improved by p53 knockdown in ATM-deficient cells or in conjunction with the specific ATM inhibitor KU-55933. AstraZeneca’s AZD6738 is a second ATR inhibitor now in clinical development. AZD6738 is an analogue of AZ20, a strong and selective ATR inhibitor that has been demonstrated to have significant single agent activity in vivo at well-tolerated doses in MRE 11A-deficient LVO xenografts [42,146][40][108]. AZD6738 possesses significantly improved solubility, bioavailability, and pharmacokinetic properties compared to AZ20 and is suitable for oral dosing [140][106]. In vitro, it suppresses the phosphorylation of the ATR downstream target CHK1 while boosting the phosphorylation of the DNA DSB marker γH2AX. The combination of this compound with carboplatin or IR has shown significantly increased antitumor growth inhibitory activity in in vivo studies. AZD6738 also showed single-agent anti-tumor efficacy in ATM-deficient but not ATM-proficient xenograft mice [136,147][103][109]. This anti-tumor effect was linked to a sustained rise in γH2AX staining in tumor tissue but only a transient increase in normal tissues such as bone marrow or the gut. This shows that a positive therapeutic index might be obtained, which is optimistic for the future clinical development of this drug.8. ATR Isomerization Potential Therapeutic Target
The most commonly used ATR inhibitors in cancer clinical studies are specific inhibitors of the enzyme ATR kinase, which plays a key role in the DNA damage checkpoint function of ATR in the nucleus. These inhibitors have no effect on cis-ATR’s antiapoptotic activity, since the new inhibitor of cis-ATR at mitochondria, which is independent from ATR kinase activation [63][54], is not affected. Such a protein target that is novel and effective in the treatment of cancer could be cis-ATR (ATR-H), potentially. Cis-ATR is not, as a matter of principle, mutagenic, but it allows cancer cells to evade apoptotic activation, which is particularly important for carcinogenesis. Cancerous cells may be resistant to death because they contain proportionally more cytoplasmic cis-ATR than healthy cells, especially when exposed to chemo- or radiotherapy, or because they have less Pin1 or less Ser428 phosphorylation in ATR [148][110]. In support, reduced levels of pSer428 ATR in the cytoplasm of advanced epithelial ovarian cancer cells are correlated with poor prognosis [149][111]. As a result, using irradiation or chemotherapy to target cis-ATR as an adjuvant treatment for cancer should preferentially kill cis-ATR-dependent cancer cells while having little influence on the normal activities of nuclear trans-ATR in cells. To guarantee cellular survival and normality, ATR is a crucial protein [31][30] that consists of cis and trans isomers that are normally active but exist in a fragile balance.9. Conclusions
The cellular functions of ATR are supported by two major activities: its kinase activity, and kinase-independent antiapoptotic activity directly at the mitochondria. ATR functions as a basal kinase and is an important part of repairing DNA, regulating the cell cycle, and apoptosis. It has a wide variety of therapeutic functions. In conclusion, ATR kinase has emerged as a promising target for cancer therapy, and the area of ATR inhibition (ATRi) is quickly expanding, with multiple early-phase clinical studies now underway.References
- Shechter, D.; Costanzo, V.; Gautier, J. Regulation of DNA replication by ATR: Signaling in response to DNA intermediates. DNA Repair. 2004, 3, 901–908.
- Zou, L.; Elledge, S.J. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science 2003, 300, 1542–1548.
- Biswas, H.; Goto, G.; Wang, W.; Sung, P.; Sugimoto, K. Ddc2ATRIP promotes Mec1ATR activation at RPA-ssDNA tracts. PLoS Genet. 2019, 15, e1008294.
- Cimprich, K.A.; Cortez, D. ATR: An essential regulator of genome integrity. Nat. Rev. Mol. Cell. Biol. 2008, 9, 616–627.
- Cortez, D.; Guntuku, S.; Qin, J.; Elledge, S.J. ATR and ATRIP: Partners in Checkpoint Signaling. Science 2001, 294, 1713–1716.
- Burrows, A.E.; Elledge, S.J. How ATR turns on: TopBP1 goes on ATRIP with ATR. Genes. Dev. 2008, 22, 1416–1421.
- Yazinski, S.A.; Zou, L. Functions, Regulation, and Therapeutic Implications of the ATR Checkpoint Pathway. Annu. Rev. Genet. 2016, 50, 155–173.
- Saldivar, J.C.; Cortez, D.; Cimprich, K.A. The essential kinase ATR: Ensuring faithful duplication of a challenging genome. Nat. Rev. Mol. Cell. Biol. 2017, 18, 622–636.
- Wu, X.; Shell, S.M.; Zou, Y. Interaction and colocalization of Rad9/Rad1/Hus1 checkpoint complex with replication protein A in human cells. Oncogene 2005, 24, 4728–4735.
- Kumagai, A.; Lee, J.; Yoo, H.Y.; Dunphy, W.G. TopBP1 Activates the ATR-ATRIP Complex. Cell 2006, 124, 943–955.
- Bass, T.E.; Luzwick, J.W.; Kavanaugh, G.; Carroll, C.; Dungrawala, H.; Glick, G.G.; Feldkamp, M.D.; Putney, R.; Chazin, W.J.; Cortez, D. ETAA1 acts at stalled replication forks to maintain genome integrity. Nat. Cell Biol. 2016, 18, 1185–1195.
- Haahr, P.; Hoffmann, S.; Tollenaere, M.A.X.; Ho, T.; Toledo, L.I.; Mann, M.; Bekker-Jensen, S.; Räschle, M.; Mailand, N. Activation of the ATR kinase by the RPA-binding protein ETAA1. Nat. Cell Biol. 2016, 18, 1196–1207.
- Sancar, A.; Laura, A.; Boltz, L.; Ünsal-Kaçmaz, K.; Linn, S. Molecular Mechanisms of Mammalian DNA Repair and the DNA Damage Checkpoints. Annu. Rev. Biochem. 2004, 73, 39–85.
- Mordes, D.A.; Glick, G.G.; Zhao, R.; Cortez, D. TopBP1 activates ATR through ATRIP and a PIKK regulatory domain. Genes. Dev. 2008, 22, 1478–1489.
- Mordes, D.A.; Cortez, D. Activation of ATR and related PIKKs. Cell. Cycle 2008, 7, 2809–2812.
- Ciccia, A.; Elledge, S.J. The DNA Damage Response: Making It Safe to Play with Knives. Mol. Cell 2010, 40, 179–204.
- Nam, E.A.; Cortez, D. ATR signalling: More than meeting at the fork. Biochem. J. 2011, 436, 527–536.
- Ma, K.; Wu, H.; Li, P.; Li, B. LC3-II may mediate ATR-induced mitophagy in dopaminergic neurons through SQSTM1/p62 pathway. Acta Biochim. Biophys. Sin. 2018, 50, 1047–1061.
- Lam, M.H.; Rosen, J.M. Chk1 versus Cdc25: Chking one’s levels of cellular proliferation. Cell Cycle 2004, 3, 1355–1357.
- Ammazzalorso, F.; Pirzio, L.M.; Bignami, M.; Franchitto, A.; Pichierri, P. ATR and ATM differently regulate WRN to prevent DSBs at stalled replication forks and promote replication fork recovery. EMBO J. 2010, 29, 3156–3169.
- Couch, F.B.; Bansbach, C.E.; Driscoll, R.; Luzwick, J.W.; Glick, G.G.; Bétous, R.; Carroll, C.M.; Jung, S.Y.; Qin, J.; Cimprich, K.A.; et al. ATR phosphorylates SMARCAL1 to prevent replication fork collapse. Genes. Dev. 2013, 27, 1610–1623.
- Chen, Y.H.; Jones, M.J.; Yin, Y.; Crist, S.B.; Colnaghi, L.; Sims, R.J., 3rd; Rothenberg, E.; Jallepalli, P.V.; Huang, T.T. ATR-mediated phosphorylation of FANCI regulates dormant origin firing in response to replication stress. Mol. Cell 2015, 58, 323–338.
- Wang, H.; Wang, H.; Powell, S.N.; Iliakis, G.; Wang, Y. ATR affecting cell radiosensitivity is dependent on homologous recombination repair but independent of nonhomologous end joining. Cancer Res. 2004, 64, 7139–7143.
- Andreassen, P.R.; D’Andrea, A.D.; Taniguchi, T. ATR couples FANCD2 monoubiquitination to the DNA-damage response. Genes. Dev. 2004, 18, 1958–1963.
- Wu, X.; Shell, S.M.; Liu, Y.; Zou, Y. ATR-dependent checkpoint modulates XPA nuclear import in response to UV irradiation. Oncogene 2007, 26, 757–764.
- Wu, X.; Shell, S.M.; Yang, Z.; Zou, Y. Phosphorylation of nucleotide excision repair factor xeroderma pigmentosum group A by ataxia telangiectasia mutated and Rad3-related-dependent checkpoint pathway promotes cell survival in response to UV irradiation. Cancer Res. 2006, 66, 2997–3005.
- Lecona, E.; Fernández-Capetillo, O. Replication stress and cancer: It takes two to tango. Exp. Cell Res. 2014, 329, 26–34.
- Brown, E.J.; Baltimore, D. ATR disruption leads to chromosomal fragmentation and early embryonic lethality. Genes. Dev. 2000, 14, 397–402.
- Menolfi, D.; Jiang, W.; Lee, B.J.; Moiseeva, T.; Shao, Z.; Estes, V.; Frattini, M.G.; Bakkenist, C.J.; Zha, S. Kinase-dead ATR differs from ATR loss by limiting the dynamic exchange of ATR and RPA. Nat. Commun. 2018, 9, 5351.
- Ruzankina, Y.; Pinzon-Guzman, C.; Asare, A.; Ong, T.; Pontano, L.; Cotsarelis, G.; Zediak, V.P.; Velez, M.; Bhandoola, A.; Brown, E.J. Deletion of the developmentally essential gene ATR in adult mice leads to age-related phenotypes and stem cell loss. Cell Stem Cell 2007, 1, 113–126.
- Schoppy, D.W.; Ragland, R.L.; Gilad, O.; Shastri, N.; Peters, A.A.; Murga, M.; Fernandez-Capetillo, O.; Diehl, J.A.; Brown, E.J. Oncogenic stress sensitizes murine cancers to hypomorphic suppression of ATR. J. Clin. Investig. 2012, 122, 241–252.
- O’Driscoll, M.; Ruiz-Perez, V.L.; Woods, C.G.; Jeggo, P.A.; Goodship, J.A. A splicing mutation affecting expression of ataxia-telangiectasia and Rad3-related protein (ATR) results in Seckel syndrome. Nat. Genet. 2003, 33, 497–501.
- Toledo, L.I.; Murga, M.; Fernandez-Capetillo, O. Targeting ATR and Chk1 kinases for cancer treatment: A new model for new (and old) drugs. Mol. Oncol. 2011, 5, 368–373.
- Höglund, A.; Nilsson, L.M.; Muralidharan, S.V.; Hasvold, L.A.; Merta, P.; Rudelius, M.; Nikolova, V.; Keller, U.; Nilsson, J.A. Therapeutic implications for the induced levels of Chk1 in Myc-expressing cancer cells. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2011, 17, 7067–7079.
- Murga, M.; Campaner, S.; Lopez-Contreras, A.J.; Toledo, L.I.; Soria, R.; Montaña, M.F.; Artista, L.; Schleker, T.; Guerra, C.; Garcia, E.; et al. Exploiting oncogene-induced replicative stress for the selective killing of Myc-driven tumors. Nat. Struct. Mol. Biol. 2011, 18, 1331–1335.
- Toledo, L.I.; Murga, M.; Zur, R.; Soria, R.; Rodriguez, A.; Martinez, S.; Oyarzabal, J.; Pastor, J.; Bischoff, J.R.; Fernandez-Capetillo, O. A cell-based screen identifies ATR inhibitors with synthetic lethal properties for cancer-associated mutations. Nat. Struct. Mol. Biol. 2011, 18, 721–727.
- Gilad, O.; Nabet, B.Y.; Ragland, R.L.; Schoppy, D.W.; Smith, K.D.; Durham, A.C.; Brown, E.J. Combining ATR suppression with oncogenic Ras synergistically increases genomic instability, causing synthetic lethality or tumorigenesis in a dosage-dependent manner. Cancer Res. 2010, 70, 9693–9702.
- Ferrao, P.T.; Bukczynska, E.P.; Johnstone, R.W.; McArthur, G.A. Efficacy of CHK inhibitors as single agents in MYC-driven lymphoma cells. Oncogene 2012, 31, 1661–1672.
- Walton, M.I.; Eve, P.D.; Hayes, A.; Valenti, M.R.; De Haven Brandon, A.K.; Box, G.; Hallsworth, A.; Smith, E.L.; Boxall, K.J.; Lainchbury, M.; et al. CCT244747 is a novel potent and selective CHK1 inhibitor with oral efficacy alone and in combination with genotoxic anticancer drugs. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2012, 18, 5650–5661.
- Cole, K.A.; Huggins, J.; Laquaglia, M.; Hulderman, C.E.; Russell, M.R.; Bosse, K.; Diskin, S.J.; Attiyeh, E.F.; Sennett, R.; Norris, G.; et al. RNAi screen of the protein kinome identifies checkpoint kinase 1 (CHK1) as a therapeutic target in neuroblastoma. Proc. Natl. Acad. Sci. USA 2011, 108, 3336–3341.
- Fokas, E.; Prevo, R.; Hammond, E.M.; Brunner, T.B.; McKenna, W.G.; Muschel, R.J. Targeting ATR in DNA damage response and cancer therapeutics. Cancer Treat. Rev. 2014, 40, 109–117.
- Foote, K.M.; Blades, K.; Cronin, A.; Fillery, S.; Guichard, S.S.; Hassall, L.; Hickson, I.; Jacq, X.; Jewsbury, P.J.; McGuire, T.M.; et al. Discovery of 4--1H-indole (AZ20): A potent and selective inhibitor of ATR protein kinase with monotherapy in vivo antitumor activity. J. Med. Chem. 2013, 56, 2125–2138.
- Reaper, P.M.; Griffiths, M.R.; Long, J.M.; Charrier, J.D.; Maccormick, S.; Charlton, P.A.; Golec, J.M.; Pollard, J.R. Selective killing of ATM- or p53-deficient cancer cells through inhibition of ATR. Nat. Chem. Biol. 2011, 7, 428–430.
- Sultana, R.; Abdel-Fatah, T.; Perry, C.; Moseley, P.; Albarakti, N.; Mohan, V.; Seedhouse, C.; Chan, S.; Madhusudan, S. Ataxia telangiectasia mutated and Rad3 related (ATR) protein kinase inhibition is synthetically lethal in XRCC1 deficient ovarian cancer cells. PLoS ONE 2013, 8, e57098.
- Mohni, K.N.; Kavanaugh, G.M.; Cortez, D. ATR pathway inhibition is synthetically lethal in cancer cells with ERCC1 deficiency. Cancer Res. 2014, 74, 2835–2845.
- Mohni, K.N.; Thompson, P.S.; Luzwick, J.W.; Glick, G.G.; Pendleton, C.S.; Lehmann, B.D.; Pietenpol, J.A.; Cortez, D. A Synthetic Lethal Screen Identifies DNA Repair Pathways that Sensitize Cancer Cells to Combined ATR Inhibition and Cisplatin Treatments. PLoS ONE 2015, 10, e0125482.
- Peasland, A.; Wang, L.Z.; Rowling, E.; Kyle, S.; Chen, T.; Hopkins, A.; Cliby, W.A.; Sarkaria, J.; Beale, G.; Edmondson, R.J.; et al. Identification and evaluation of a potent novel ATR inhibitor, NU6027, in breast and ovarian cancer cell lines. Br. J. Cancer 2011, 105, 372–381.
- Bristow, R.G.; Hill, R.P. Hypoxia and metabolism. Hypoxia, DNA repair and genetic instability. Nat. Rev. Cancer 2008, 8, 180–192.
- Hammond, E.M.; Dorie, M.J.; Giaccia, A.J. Inhibition of ATR leads to increased sensitivity to hypoxia/reoxygenation. Cancer Res. 2004, 64, 6556–6562.
- Pires, I.M.; Olcina, M.M.; Anbalagan, S.; Pollard, J.R.; Reaper, P.M.; Charlton, P.A.; McKenna, W.G.; Hammond, E.M. Targeting radiation-resistant hypoxic tumour cells through ATR inhibition. Br. J. Cancer 2012, 107, 291–299.
- Prevo, R.; Fokas, E.; Reaper, P.M.; Charlton, P.A.; Pollard, J.R.; McKenna, W.G.; Muschel, R.J.; Brunner, T.B. The novel ATR inhibitor VE-821 increases sensitivity of pancreatic cancer cells to radiation and chemotherapy. Cancer Biol. 2012, 13, 1072–1081.
- Pires, I.M.; Bencokova, Z.; Milani, M.; Folkes, L.K.; Li, J.L.; Stratford, M.R.; Harris, A.L.; Hammond, E.M. Effects of acute versus chronic hypoxia on DNA damage responses and genomic instability. Cancer Res. 2010, 70, 925–935.
- Flynn, R.L.; Cox, K.E.; Jeitany, M.; Wakimoto, H.; Bryll, A.R.; Ganem, N.J.; Bersani, F.; Pineda, J.R.; Suvà, M.L.; Benes, C.H.; et al. Alternative lengthening of telomeres renders cancer cells hypersensitive to ATR inhibitors. Science 2015, 347, 273–277.
- Hilton, B.A.; Li, Z.; Musich, P.R.; Wang, H.; Cartwright, B.M.; Serrano, M.; Zhou, X.Z.; Lu, K.P.; Zou, Y. ATR Plays a Direct Antiapoptotic Role at Mitochondria, which Is Regulated by Prolyl Isomerase Pin1. Mol. Cell 2015, 60, 35–46.
- Makinwa, Y.; Cartwright, B.M.; Musich, P.R.; Li, Z.; Biswas, H.; Zou, Y. PP2A Regulates Phosphorylation-Dependent Isomerization of Cytoplasmic and Mitochondrial-Associated ATR by Pin1 in DNA Damage Responses. Front. Cell Dev. Biol. 2020, 8, 813.
- Murga, M.; Bunting, S.; Montaña, M.F.; Soria, R.; Mulero, F.; Cañamero, M.; Lee, Y.; McKinnon, P.J.; Nussenzweig, A.; Fernandez-Capetillo, O. A mouse model of ATR-Seckel shows embryonic replicative stress and accelerated aging. Nat. Genet. 2009, 41, 891–898.
- Biswas, H.; Zhao, S.J.; Makinwa, Y.; Bassett, J.S.; Musich, P.R.; Liu, J.Y.; Zou, Y. Prolyl Isomerization-Mediated Conformational Changes Define ATR Subcellular Compartment-Specific Functions. Front. Cell. Dev. Biol. 2022, 10, 826576.
- Musich, P.R.; Li, Z.; Zou, Y. Xeroderma Pigmentosa Group A (XPA), Nucleotide Excision Repair and Regulation by ATR in Response to Ultraviolet Irradiation. Adv. Exp. Med. Biol. 2017, 996, 41–54.
- Makinwa, Y.; Musich, P.R.; Zou, Y. Phosphorylation-Dependent Pin1 Isomerization of ATR: Its Role in Regulating ATR’s Anti-apoptotic Function at Mitochondria, and the Implications in Cancer. Front. Cell. Dev. Biol. 2020, 8, 281.
- Cho, S.; Irianto, J.; Discher, D.E. Mechanosensing by the nucleus: From pathways to scaling relationships. J. Cell Biol. 2017, 216, 305–315.
- Kirby, T.J.; Lammerding, J. Emerging views of the nucleus as a cellular mechanosensor. Nat. Cell Biol. 2018, 20, 373–381.
- Kidiyoor, G.R.; Li, Q.; Bastianello, G.; Bruhn, C.; Giovannetti, I.; Mohamood, A.; Beznoussenko, G.V.; Mironov, A.; Raab, M.; Piel, M.; et al. ATR is essential for preservation of cell mechanics and nuclear integrity during interstitial migration. Nat. Commun. 2020, 11, 4828.
- Tan, X.; Zhou, F.; Wan, J.; Hang, J.; Chen, Z.; Li, B.; Zhang, C.; Shao, K.; Jiang, P.; Shi, S.; et al. Pin1 expression contributes to lung cancer: Prognosis and carcinogenesis. Cancer Biol. 2010, 9, 111–119.
- Liu, M.; Zeng, T.; Zhang, X.; Liu, C.; Wu, Z.; Yao, L.; Xie, C.; Xia, H.; Lin, Q.; Xie, L.; et al. ATR/Chk1 signaling induces autophagy through sumoylated RhoB-mediated lysosomal translocation of TSC2 after DNA damage. Nat. Commun. 2018, 9, 4139.
- Kang, C.; Xu, Q.; Martin, T.D.; Li, M.Z.; Demaria, M.; Aron, L.; Lu, T.; Yankner, B.A.; Campisi, J.; Elledge, S.J. The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4. Science 2015, 349, aaa5612.
- Ayala, G.; Wang, D.; Wulf, G.; Frolov, A.; Li, R.; Sowadski, J.; Wheeler, T.M.; Lu, K.P.; Bao, L. The prolyl isomerase Pin1 is a novel prognostic marker in human prostate cancer. Cancer Res. 2003, 63, 6244–6251.
- He, J.; Zhou, F.; Shao, K.; Hang, J.; Wang, H.; Rayburn, E.; Xiao, Z.X.; Lee, S.W.; Xue, Q.; Feng, X.L.; et al. Overexpression of Pin1 in non-small cell lung cancer (NSCLC) and its correlation with lymph node metastases. Lung Cancer 2007, 56, 51–58.
- Girardini, J.E.; Napoli, M.; Piazza, S.; Rustighi, A.; Marotta, C.; Radaelli, E.; Capaci, V.; Jordan, L.; Quinlan, P.; Thompson, A.; et al. A Pin1/mutant p53 axis promotes aggressiveness in breast cancer. Cancer Cell 2011, 20, 79–91.
- Luo, M.L.; Gong, C.; Chen, C.H.; Lee, D.Y.; Hu, H.; Huang, P.; Yao, Y.; Guo, W.; Reinhardt, F.; Wulf, G.; et al. Prolyl isomerase Pin1 acts downstream of miR200c to promote cancer stem-like cell traits in breast cancer. Cancer Res. 2014, 74, 3603–3616.
- Rustighi, A.; Zannini, A.; Tiberi, L.; Sommaggio, R.; Piazza, S.; Sorrentino, G.; Nuzzo, S.; Tuscano, A.; Eterno, V.; Benvenuti, F.; et al. Prolyl-isomerase Pin1 controls normal and cancer stem cells of the breast. EMBO Mol. Med. 2014, 6, 99–119.
- Xu, M.; Cheung, C.C.; Chow, C.; Lun, S.W.; Cheung, S.T.; Lo, K.W. Overexpression of PIN1 Enhances Cancer Growth and Aggressiveness with Cyclin D1 Induction in EBV-Associated Nasopharyngeal Carcinoma. PLoS ONE 2016, 11, e0156833.
- Nakatsu, Y.; Yamamotoya, T.; Ueda, K.; Ono, H.; Inoue, M.K.; Matsunaga, Y.; Kushiyama, A.; Sakoda, H.; Fujishiro, M.; Matsubara, A.; et al. Prolyl isomerase Pin1 in metabolic reprogramming of cancer cells. Cancer Lett. 2020, 470, 106–114.
- Chen, Y.; Wu, Y.R.; Yang, H.Y.; Li, X.Z.; Jie, M.M.; Hu, C.J.; Wu, Y.Y.; Yang, S.M.; Yang, Y.B. Prolyl isomerase Pin1: A promoter of cancer and a target for therapy. Cell Death Dis. 2018, 9, 883.
- Hennig, L.; Christner, C.; Kipping, M.; Schelbert, B.; Rucknagel, K.P.; Grabley, S.; Kullertz, G.; Fischer, G. Selective inactivation of parvulin-like peptidyl-prolyl cis/trans isomerases by juglone. Biochemistry 1998, 37, 5953–5960.
- Campaner, E.; Rustighi, A.; Zannini, A.; Cristiani, A.; Piazza, S.; Ciani, Y.; Kalid, O.; Golan, G.; Baloglu, E.; Shacham, S.; et al. A covalent PIN1 inhibitor selectively targets cancer cells by a dual mechanism of action. Nat. Commun. 2017, 8, 15772.
- Estey, E.; Thall, P.F.; Pierce, S.; Kantarjian, H.; Keating, M. Treatment of newly diagnosed acute promyelocytic leukemia without cytarabine. J. Clin. Oncol. 1997, 15, 483–490.
- Budd, G.T.; Adamson, P.C.; Gupta, M.; Homayoun, P.; Sandstrom, S.K.; Murphy, R.F.; McLain, D.; Tuason, L.; Peereboom, D.; Bukowski, R.M.; et al. Phase I/II trial of all-trans retinoic acid and tamoxifen in patients with advanced breast cancer. Clin. Cancer Res. 1998, 4, 635–642.
- Shen, Z.X.; Shi, Z.Z.; Fang, J.; Gu, B.W.; Li, J.M.; Zhu, Y.M.; Shi, J.Y.; Zheng, P.Z.; Yan, H.; Liu, Y.F.; et al. All-trans retinoic acid/As2O3 combination yields a high quality remission and survival in newly diagnosed acute promyelocytic leukemia. Proc. Natl. Acad. Sci. USA 2004, 101, 5328–5335.
- Lu, Z.; Hunter, T. Prolyl isomerase Pin1 in cancer. Cell Res. 2014, 24, 1033–1049.
- Wei, S.; Kozono, S.; Kats, L.; Nechama, M.; Li, W.; Guarnerio, J.; Luo, M.; You, M.H.; Yao, Y.; Kondo, A.; et al. Active Pin1 is a key target of all-trans retinoic acid in acute promyelocytic leukemia and breast cancer. Nat. Med. 2015, 21, 457–466.
- Zhou, X.Z.; Lu, K.P. The isomerase PIN1 controls numerous cancer-driving pathways and is a unique drug target. Nat. Rev. Cancer 2016, 16, 463–478.
- Lian, X.; Lin, Y.M.; Kozono, S.; Herbert, M.K.; Li, X.; Yuan, X.; Guo, J.; Guo, Y.; Tang, M.; Lin, J.; et al. Pin1 inhibition exerts potent activity against acute myeloid leukemia through blocking multiple cancer-driving pathways. J. Hematol. Oncol. 2018, 11, 73.
- Muindi, J.; Frankel, S.R.; Miller, W.H., Jr.; Jakubowski, A.; Scheinberg, D.A.; Young, C.W.; Dmitrovsky, E.; Warrell, R.P., Jr. Continuous treatment with all-trans retinoic acid causes a progressive reduction in plasma drug concentrations: Implications for relapse and retinoid “resistance” in patients with acute promyelocytic leukemia. Blood 1992, 79, 299–303.
- Decensi, A.; Robertson, C.; Guerrieri-Gonzaga, A.; Serrano, D.; Cazzaniga, M.; Mora, S.; Gulisano, M.; Johansson, H.; Galimberti, V.; Cassano, E.; et al. Randomized double-blind 2 × 2 trial of low-dose tamoxifen and fenretinide for breast cancer prevention in high-risk premenopausal women. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2009, 27, 3749–3756.
- Arrieta, O.; Gonzalez-De la Rosa, C.H.; Arechaga-Ocampo, E.; Villanueva-Rodriguez, G.; Ceron-Lizarraga, T.L.; Martinez-Barrera, L.; Vazquez-Manriquez, M.E.; Rios-Trejo, M.A.; Alvarez-Avitia, M.A.; Hernandez-Pedro, N.; et al. Randomized phase II trial of All-trans-retinoic acid with chemotherapy based on paclitaxel and cisplatin as first-line treatment in patients with advanced non-small-cell lung cancer. J. Clin. Oncol. 2010, 28, 3463–3471.
- Moore, J.D.; Potter, A. Pin1 inhibitors: Pitfalls, progress and cellular pharmacology. Bioorg. Med. Chem. Lett. 2013, 23, 4283–4291.
- Han, H.J.; Choi, B.Y.; Surh, Y.J. Dual Roles of Pin1 in Cancer Development and Progression. Curr. Pharm. Des. 2017, 23, 4422–4425.
- Weber, A.M.; Ryan, A.J. ATM and ATR as therapeutic targets in cancer. Pharmacol. Ther. 2015, 149, 124–138.
- Ma, C.X.; Janetka, J.W.; Piwnica-Worms, H. Death by releasing the breaks: CHK1 inhibitors as cancer therapeutics. Trends Mol. Med. 2011, 17, 88–96.
- Vance, S.; Liu, E.; Zhao, L.; Parsels, J.D.; Parsels, L.A.; Brown, J.L.; Maybaum, J.; Lawrence, T.S.; Morgan, M.A. Selective radiosensitization of p53 mutant pancreatic cancer cells by combined inhibition of Chk1 and PARP1. Cell Cycle 2011, 10, 4321–4329.
- Dent, P.; Tang, Y.; Yacoub, A.; Dai, Y.; Fisher, P.B.; Grant, S. CHK1 inhibitors in combination chemotherapy: Thinking beyond the cell cycle. Mol. Interv. 2011, 11, 133–140.
- Bunch, R.T.; Eastman, A. Enhancement of cisplatin-induced cytotoxicity by 7-hydroxystaurosporine (UCN-01), a new G2-checkpoint inhibitor. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 1996, 2, 791–797.
- Matthews, D.J.; Yakes, F.M.; Chen, J.; Tadano, M.; Bornheim, L.; Clary, D.O.; Tai, A.; Wagner, J.M.; Miller, N.; Kim, Y.D.; et al. Pharmacological abrogation of S-phase checkpoint enhances the anti-tumor activity of gemcitabine in vivo. Cell Cycle 2007, 6, 104–110.
- Sarkaria, J.N.; Busby, E.C.; Tibbetts, R.S.; Roos, P.; Taya, Y.; Karnitz, L.M.; Abraham, R.T. Inhibition of ATM and ATR kinase activities by the radiosensitizing agent, caffeine. Cancer Res. 1999, 59, 4375–4382.
- Blasina, A.; Price, B.D.; Turenne, G.A.; McGowan, C.H. Caffeine inhibits the checkpoint kinase ATM. Curr. Biol. CB 1999, 9, 1135–1138.
- Charrier, J.D.; Durrant, S.J.; Golec, J.M.; Kay, D.P.; Knegtel, R.M.; MacCormick, S.; Mortimore, M.; O’Donnell, M.E.; Pinder, J.L.; Reaper, P.M.; et al. Discovery of potent and selective inhibitors of ataxia telangiectasia mutated and Rad3 related (ATR) protein kinase as potential anticancer agents. J. Med. Chem. 2011, 54, 2320–2330.
- Fokas, E.; Prevo, R.; Pollard, J.R.; Reaper, P.M.; Charlton, P.A.; Cornelissen, B.; Vallis, K.A.; Hammond, E.M.; Olcina, M.M.; Gillies McKenna, W.; et al. Targeting ATR in vivo using the novel inhibitor VE-822 results in selective sensitization of pancreatic tumors to radiation. Cell. Death Dis. 2012, 3, e441.
- Huntoon, C.J.; Flatten, K.S.; Wahner Hendrickson, A.E.; Huehls, A.M.; Sutor, S.L.; Kaufmann, S.H.; Karnitz, L.M. ATR inhibition broadly sensitizes ovarian cancer cells to chemotherapy independent of BRCA status. Cancer Res. 2013, 73, 3683–3691.
- Vávrová, J.; Zárybnická, L.; Lukášová, E.; Řezáčová, M.; Novotná, E.; Sinkorová, Z.; Tichý, A.; Pejchal, J.; Durišová, K. Inhibition of ATR kinase with the selective inhibitor VE-821 results in radiosensitization of cells of promyelocytic leukaemia (HL-60). Radiat. Environ. Biophys. 2013, 52, 471–479.
- Jossé, R.; Martin, S.E.; Guha, R.; Ormanoglu, P.; Pfister, T.D.; Reaper, P.M.; Barnes, C.S.; Jones, J.; Charlton, P.; Pollard, J.R.; et al. ATR inhibitors VE-821 and VX-970 sensitize cancer cells to topoisomerase i inhibitors by disabling DNA replication initiation and fork elongation responses. Cancer Res. 2014, 74, 6968–6979.
- Hall, A.B.; Newsome, D.; Wang, Y.; Boucher, D.M.; Eustace, B.; Gu, Y.; Hare, B.; Johnson, M.A.; Milton, S.; Murphy, C.E.; et al. Potentiation of tumor responses to DNA damaging therapy by the selective ATR inhibitor VX-970. Oncotarget 2014, 5, 5674–5685.
- Abdel-Fatah, T.M.; Middleton, F.K.; Arora, A.; Agarwal, D.; Chen, T.; Moseley, P.M.; Perry, C.; Doherty, R.; Chan, S.; Green, A.R.; et al. Untangling the ATR-CHEK1 network for prognostication, prediction and therapeutic target validation in breast cancer. Mol. Oncol. 2015, 9, 569–585.
- Jones, C.D.; Blades, K.; Foote, K.M.; Guichard, S.M.; Jewsbury, P.J.; McGuire, T.; Nissink, J.W.; Odedra, R.; Tam, K.; Thommes, P. Discovery of AZD6738, a potent and selective inhibitor with the potential to test the clinical efficacy of ATR kinase inhibition in cancer patients. Cancer Res. 2013, 73, 2348.
- Clack, G.; Lau, A.; Pierce, A.; Smith, S.; Stephens, C. ATR inhibitor AZD6738. Ann. Oncol. 2015, 26, ii8.
- Nishida, H.; Tatewaki, N.; Nakajima, Y.; Magara, T.; Ko, K.M.; Hamamori, Y.; Konishi, T. Inhibition of ATR protein kinase activity by schisandrin B in DNA damage response. Nucleic Acids Res. 2009, 37, 5678–5689.
- Maira, S.M.; Stauffer, F.; Brueggen, J.; Furet, P.; Schnell, C.; Fritsch, C.; Brachmann, S.; Chène, P.; De Pover, A.; Schoemaker, K.; et al. Identification and characterization of NVP-BEZ235, a new orally available dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor with potent in vivo antitumor activity. Mol. Cancer Ther. 2008, 7, 1851–1863.
- Konstantinidou, G.; Bey, E.A.; Rabellino, A.; Schuster, K.; Maira, M.S.; Gazdar, A.F.; Amici, A.; Boothman, D.A.; Scaglioni, P.P. Dual phosphoinositide 3-kinase/mammalian target of rapamycin blockade is an effective radiosensitizing strategy for the treatment of non-small cell lung cancer harboring K-RAS mutations. Cancer Res. 2009, 69, 7644–7652.
- Jacq, X.; Smith, L.; Brown, E.; Hughes, A.; Odedra, R.; Heathcote, D.; Barnes, J.; Powell, S.; Maguire, S.; Pearson, V.; et al. Abstract 1823: AZ20, a novel potent and selective inhibitor of ATR kinase with in vivo antitumour activity. Cancer Res. 2012, 72, 1823.
- Guichard, S.M.; Brown, E.; Odedra, R.; Hughes, A.; Heathcote, D.; Barnes, J.; Lau, A.; Powell, S.; Jones, C.D.; Nissink, W.; et al. Abstract 3343: The pre-clinical in vitro and in vivo activity of AZD6738: A potent and selective inhibitor of ATR kinase. Cancer Res. 2013, 73, 3343.
- Ibarra, M.S.; Borini Etichetti, C.; Di Benedetto, C.; Rosano, G.L.; Margarit, E.; Del Sal, G.; Mione, M.; Girardini, J. Dynamic regulation of Pin1 expression and function during zebrafish development. PLoS ONE 2017, 12, e0175939.
- Lee, B.; Lee, H.J.; Cho, H.Y.; Suh, D.H.; Kim, K.; No, J.H.; Kim, H.; Kim, Y.B. Ataxia-Telangiectasia and RAD3-Related and Ataxia-Telangiectasia-Mutated Proteins in Epithelial Ovarian Carcinoma: Their Expression and Clinical Significance. Anticancer Res. 2015, 35, 3909–3916.
More