Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Rita Xu and Version 1 by Anna Kakehashi.
Non-alcoholic fatty liver disease (NAFLD) or metabolic dysfunction-associated steatotic liver disease (MASLD) and steatohepatitis (NASH) are chronic hepatic conditions leading to hepatocellular carcinoma (HCC) development. According to the recent “multiple-parallel-hits hypothesis”, NASH could be caused by abnormal metabolism, accumulation of lipids, mitochondrial dysfunction, and oxidative and endoplasmic reticulum stresses and is found in obese and non-obese patients.
- biomarker
- hepatocarcinogenesis mechanisms
- NAFLD
1. Introduction
The global epidemic of obesity and metabolic syndrome has become a significant threat to human health worldwide. Recent epidemiological evidence shows that non-alcoholic fatty liver disease (NAFLD), recently termed metabolic dysfunction-associated steatotic liver disease (MASLD), and steatohepatitis (NASH) have rapidly become leading etiologies predisposing to fibrosis, cirrhosis, and hepatocellular carcinoma (HCC) development due to the rapid increase in metabolic syndrome and obesity in the past decades and newly available therapies for hepatitis C (HCV) and B virus (HBV)-associated HCC [1,2,3,4][1][2][3][4] (Figure 1). NAFLD/NASH-HCC is known to have even lower survival rates than viral hepatitis HCC and is generally developed based on metabolic syndrome, hyperinsulinemia, insulin resistance, and dyslipidemia, and is characterized by fat accumulation in the liver, either due to the increased inflow of free fatty acids or de novo lipogenesis [3,4][3][4]. NAFLD, a hepatic manifestation of insulin resistance, is very important in the pathophysiology of type 2 diabetes (T2DM). Furthermore, T2DM confers a three-fold risk of HCC [5]. However, in recent studies, it was reported to occur in cirrhosis-free and non-obese patients (“lean NAFLD”) and to be triggered by endotoxin-related inflammation in the gut and adipocytokines, leptin, and adiponectin [4,6,7,8][4][6][7][8].

Figure 1. The developmental process of NAFLD/NASH-associated HCC in human and NASH model mice. The collagen-rich fibrotic regions are visualized via Masson’s trichrome staining. Transmission electron microscopy (TEM) is performed in TSOD mice HCC. LD: lipid droplet, Mi: mitochondria, N: nuclei, ER: endoplasmic reticulum, P: peroxisome, L: lysosome.
As for the mechanism, recently, the “multiple-parallel-hits” hypothesis was proposed, in which NASH was suggested to occur upon chronic liver damage caused by abnormal metabolism and lipotoxicity, lipid peroxidation, mitochondrial dysfunction, oxidative, endoplasmic reticulum stresses, inflammation, and cell death [9,10][9][10].
Monitoring the degree of hepatic necrotizing inflammation and fibrosis is crucial to predicting the long-term prognosis of NAFLD patients. Early intervention is necessary to slow disease progression. While liver biopsy is the gold standard for diagnosis, it is not widely used due to its invasive nature. Additionally, alpha-fetoprotein (AFP) is commonly used to diagnose HCC in NASH patients, but recent studies have found it difficult to distinguish between NASH and NASH-HCC patients [11,12][11][12]. Therefore, novel biomarkers are urgently needed for accurate and early NASH-HCC diagnosis and prognosis. The emergence of genomics, transcriptomics, proteomics, metabolomics, and lipidomics provides an opportunity to gain insight into multiple processes that contribute to the development of non-alcoholic steatohepatitis (NASH). Moreover, these technologies can aid in the identification of novel biomarkers for the diagnosis and prognosis of non-alcoholic fatty liver disease (NAFLD), NASH, and hepatocellular carcinoma (HCC). On the other hand, the discovery of NAFLD/NASH-associated physiopathological signaling cascades has benefited from large-scale omics studies describing the modulation of genes, proteins, and metabolites. In addition to well-known regulators of NAFLD/NASH hepatocarcinogenesis, such as transforming growth factor beta (TGF-β), SMAD family member 3 (SMAD3), insulin-like growth factor 1 (IGF1), peroxisome proliferation activating receptors (PPARs), sterol regulatory element-binding protein and liver X receptor α (SREBP-LXRα), CCAAT/enhancer-binding protein beta (CEBPB), nuclear factor κ-light-chain-binding protein (NFkB), c-myc, Wnt/β-catenin, signal transducer and activator of transcription 3 (STAT3), and p53, recent research has highlighted the important role of protein kinases mammalian target of rapamycin (mTOR), protein receptor sequestosome 1 (SQSTM1, p62), PI3K/Akt, PKC, MAPK, ErbB, nuclear factor (erythroid-derived 2)-like 2 (Nrf2), hepatocyte nuclear factor 1α (HNF1α) and 4α (HNF4α), and nuclear-receptor-interacting protein 1 (NRIP1; nuclear factor RIP140) in this process [12,13,14,15,16,17][12][13][14][15][16][17].
2. Signaling and Metabolic Pathways in the Regulation of NAFLD/NASH Progression, Hepatocarcinogenesis, and Associated Biomarkers
2.1. mTOR Pathway as a Multifunctional Driver of Metabolic Syndrome-Associated Hepatocarcinogenesis
Numerous clinical studies have established a causal connection between metabolic syndrome and diabetes with various types of cancer, including HCC. These conditions heighten the risk of developing cancer and facilitate the process of hepatocarcinogenesis by activating inflammatory pathways that generate proinflammatory cytokines and reactive oxygen species (ROS). This, in turn, leads to genomic instability, cellular proliferation, and the suppression of apoptosis [18] (Figure 2). The accumulation of lipids in the liver is a significant contributor to the progression of NAFLD in individuals with T2DM. The mTOR signaling pathway has a crucial role in regulating metabolic processes in various organs, including hepatic lipid metabolism, insulin resistance, chronic liver inflammation, and, ultimately, the advancement of NAFLD [18].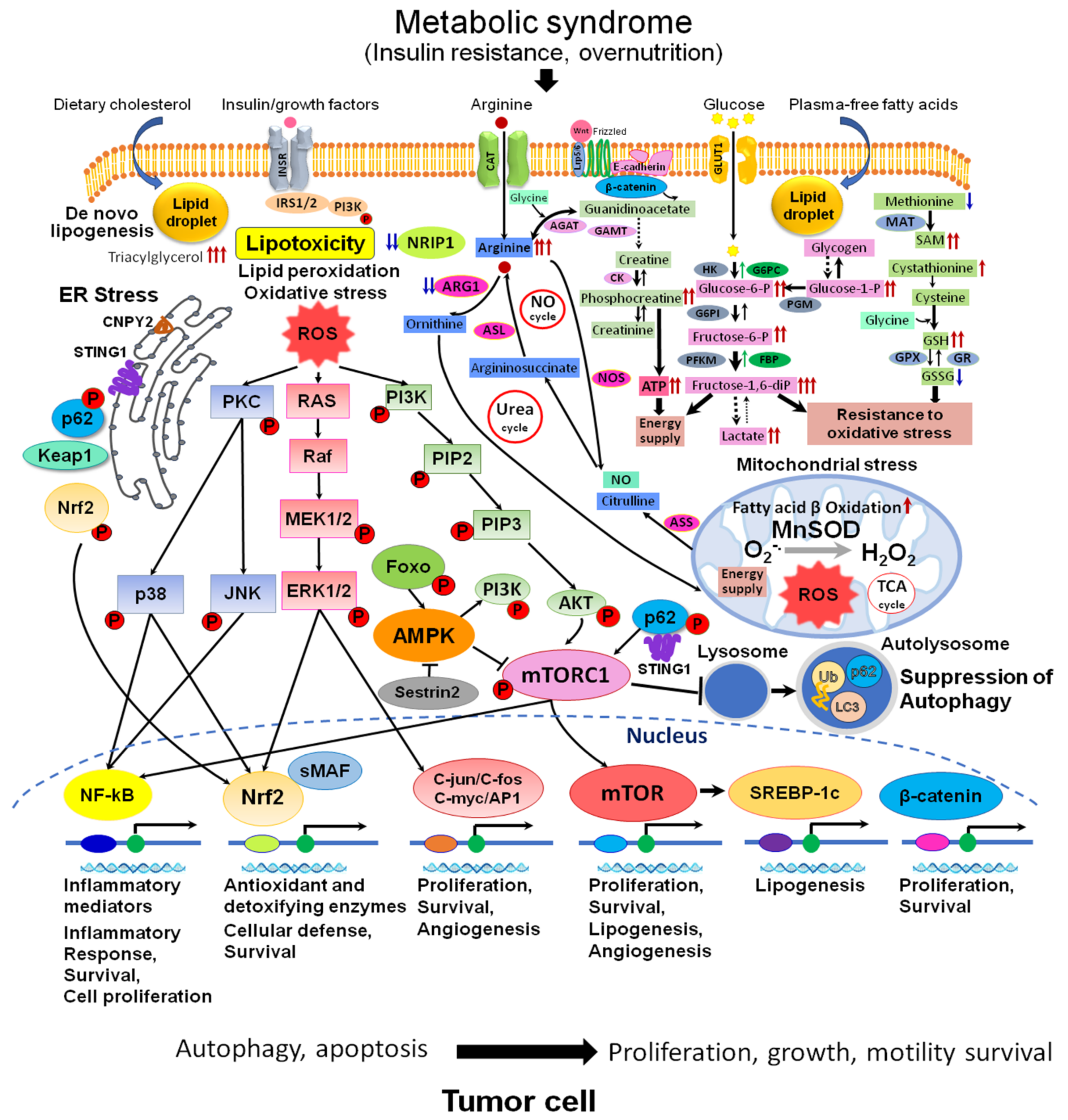
Figure 2. Metabolic and signaling pathways in NAFLD/NASH-associated HCC. Abnormal metabolism: accumulation of arginine, glucose metabolites, phosphocreatine, SAM, GSH, and alterations of urea and NO cycles are observed in NAFLD/NASH HCC. These events are associated with developed lipotoxicity, activation of de novo lipogenesis (SREBP-1), lipid peroxidation, fatty acid β oxidation, mitochondrial dysfunction, activation of oxidative (MAPK, Nrf2, NF-kB, C-jun/C-fos/C-myc/AP1) and ER stresses (p62, Keap1, Nrf2, STING, CNPY2)-related signaling, wnt/β-catenin pathway, and suppression of autophagy (mTORC1, AMPK, p62), with the central role for mTOR pathway. Abbreviations: ROS, reactive oxygen species; NO, nitric oxide; ER, endoplasmic reticulum; TCA, tricarboxylic acid cycle; INSR, insulin receptor; GLUT1, glucose trasporter 1; STING1, stimulator of interferon response cGAMP interactor 1; SAM, S-adeninemethyonine; SAH, S-adenosyl-L-homocysteine; Ub, ubiquitin; LC3, microtubule-associated protein 1A/1B−light chain 3; sMAF, small maf protein; CAT, cationic amino acid transporter; PKC, protein kinase C; MnSOD, manganese superoxide dismutase; PIP2, phosphatidylinositol 4,5-bisphosphate; PIP3, phosphatidylinositol 3,4,5-triphosphate; PI3K, phosphoinositide 3-kinase; AKT, protein kinase B (serine/threonine-specific protein kinase); p38, p38 mitogen-activated protein kinase; MEK, mitogen-activated protein kinase kinase; ERK, extracellular signal-regulated kinase; JNK, c-Jun N-terminal kinase; ARG1, arginase1; ASL, argininosuccinate lyase; ASS, argininosuccinate synthase; AGAT, arginine-glycine amidinotransferase; GAMT, guanidinoacetate N-methyltransferase; NOS, nitric oxide synthase; CK, creatine kinase; HK, hexokinase; G6PI, glucose-6-phosphate isomerase; PFKM, 6-phosphofructokinase; G6PC, glucose-6-phosphatase; FBP, fructose 1,6-bisphosphate aldolase; PGM, phosphoglycerate mutase; MAT, methionine adenosyltransferase; GPX, glutathione peroxidase, GR, glutathione reductase.
2.2. Disturbances of Metabolism in NAFLD/NASH-HCC
Untargeted metabolomics has been recently applied to find novel and peculiar metabolite profiles in NADLD/NASH/HCC patients and NASH model mice and to give insights into discovering new biomarkers in NASH pathophysiology. The liver of TSOD NASH model mice showed an increase in lipogenesis, inflammation, peroxisome proliferation, and fibrogenesis, along with a significant rise in glucose metabolites, including glucose 1-phosphate, glucose 6-phosphate, galactose 1-phosphate, fructose 6-phosphate, and primarily fructose 1,6-diphosphate, in HCCs [14] (Figure 2). It was found that TSOD mice with liver preneoplastic lesions and tumors had significantly increased levels of polysaccharides, along with a drastic increase in amino acid L-arginine and notable increases in phosphocreatine, S-adenosylmethionine/S-adenosylhomocysteine ratio, and adenylate and guanylate energy charges [14]. Masarone et al. conducted a recent human metabolomics study that analyzed 307 subjects with steatosis, NASH, and NASH-cirrhosis. Through the study, they were able to identify specific metabolites that distinguished between the different patient classes [52][27]. In this investigation, an elevation of glycocholic acid, taurocholic acid, phenylalanine, and branched-chain amino-acids accompanied by a decrease in glutathione was found with the increase in the severity of the disease from steatosis to NASH and NASH-cirrhosis. Very recently, Ahmed et al. reported a depletion of amino acid leucine in NASH-HCC as compared with NASH patients [12]. In the past, branched-chain amino acids have been effective in inhibiting the growth and angiogenesis of HCC through the suppression of insulin resistance and degradation of vascular endothelial growth factor mRNA [12,53][12][28]. Significantly, they were found to trigger apoptosis in liver cancer cells by disrupting mTOR-related pathways [54][29]. Metabolomics profiling has proven to be an effective non-invasive diagnostic tool for distinguishing between NAFLD/NASH-HCC and identifying the different stages of the disease [14,52][14][27]. The emerging area of NAFLD/NASH-associated cancer development and progression was devoted to research on lipid metabolism [55][30]. The risk of NAFLD was reported to be increased with an imbalance between fatty acid uptake, oxidation, and secretion and a high level of plasma phospholipids (e.g., phosphatidylethanolamine), which can act as signal transductors with oncogenic and tumor-suppressive roles [12]. In addition, suppression of mitochondrial β-oxidation with increased peroxisomal and microsomal ω-oxidation accompanied by accumulation of hepatic fatty acids in NASH resulted in the activation of lipotoxicity and progression of the disease. The HCCs in TSOD mice showed alterations in intracellular pathways, such as suppression of the urea cycle, methionine and putrescine degradation pathways, and an increase in cellular antioxidants like glutathione (GSH) levels. This indicates a boost in antioxidant activity in the HCCs [13]. Proteome analysis of HCCs revealed that increased levels of L-arginine significantly inhibited urea cycle enzymes, such as ARG1, argininosuccinate lyase (ASL), argininosuccinate synthase 1 (ASS1), and ornithine carbamoyltransferase (OTC). The study proposes that liver tumor-initiating cells contribute to the development of T2DM/NASH hepatocarcinogenesis by increasing glucose metabolites and L-arginine levels. This triggers glucose uptake, creatine metabolism, oxidative stress resistance, and tumor cell proliferation [14]. These findings highlighted the association between elevated L-arginine and inhibition of these enzymes.2.3. Formation of Oxidative Stress and Resistance Mechanisms
Various sources contribute to the formation of oxidative stress, including the oxidation of free fatty acids, iron overload, inflammatory cytokines, lipid peroxidation, and mitochondrial dysfunction. Oxidative stress plays a crucial role in the progression of simple steatosis to NASH and hepatocarcinogenesis [56][31]. When the balance between the production of ROS (such as hydroxyl (OH•), superoxide (O2•−) radicals, and nitric oxide (•NO) and non-radical species (H2O2), as well as the protection from oxidative damage through antioxidant enzyme systems and DNA repair, is disrupted in the liver as in NAFLD/NASH, it can lead to the onset and progression of hepatocarcinogenesis [57][32]. Excessive ROS production can harm DNA and proteins, disrupt mitochondrial conditions, trigger apoptosis, and alter cellular signaling pathways like MAPK. This can impact regenerative cell proliferation, growth, and angiogenesis [57][32]. The development of oxidative stress and its influence on hepatocarcinogenesis was studied in NASH animal models. Formation of oxidative stress and damage to liver cell DNA in terms of 8-hydroxy-2-deoxyguanosine (8-OHdG) in TSOD and fatty liver Shionogi (FLS) NASH model mice was considered a key event in the initiation of T2DM/NASH-associated hepatocarcinogenesis [14,58][14][33]. From proteomics studies, NASH-associated hepatocarcinogenesis was found to be associated with the activation of nuclear factor (erythroid-derived 2)-like 2 (Nrf2)-mediated signaling, which is known to mediate resistance to oxidative stress injury [13]. In addition, alteration of protein expression of numerous enzymes involved in the control of mitochondrial function was also detected in NASH-associated human HCCs. Human and STAM NASH model mice HCCs exhibited strong overexpression of transcriptional factors prohibitin 1 (PHB1) and prohibitin 2 (PHB2). These factors play crucial roles in controlling mitochondrial respiration, function, cell proliferation, and apoptosis [13]. In addition, the study revealed an increase in the activity of superoxide dismutase manganese [Mn], mitochondrial (SOD2), and thioredoxin (TXN), while catalase (CAT) was found to be decreased in tumors [13]. In another NASH model containing mice fed with a methionine and choline-deficient diet (MCD), proteomic analysis of livers revealed significant elevation of peroxiredoxins 1 (Prdx1) and 6 (Prdx6) in correlation with TGFβ1, TNFα, and TLR4, CYP2E1, cytokeratin 8 (CK8), cytokeratin 18 (CK18), fructose-1,6-bisphosphatase 1 and vimentin, and downregulation of proteins involved in methionine (Met) metabolism and oxidative stress in NAFLD/NASH [27][34]. From those studies, all observed metabolome and proteome alterations pointed to the formation of oxidative stress resistance in liver tumor cells as a consequence of lipotoxicity and insulin resistance. Among proteins involved in the development of liver tissue cellular defense against oxidative stress, oxidative DNA damage, inflammation, and steatohepatitis, peroxiredoxins 1 (Prx1) and 6 (Prdx6) and sestrin 2 (SESN2) were considered promising potential diagnostic biomarkers for the early stage of NAFLD/NASH, which may also influence the mTOR [27,28][34][35]. Prdx1 and Prdx6 are involved in Akt/mTOR, AMPK/mTOR, PI3K/Akt, FoxO and p53 signaling pathways, and ubiquitin-mediated proteolysis [59,60][36][37]. It was also demonstrated that protein levels of SESN2 were positively correlated with AMPK/mTOR pathway components and induced expression of lipogenesis-related genes in NAFLD model HFD-fed C57BL/6 mice liver and palmitic acid (PA)-stimulated HepG2 cells [28][35].2.4. Glycoproteins-Related Pathways in NASH HCC and Potential Biomarkers
Ramachandran et al. have revealed through their study of human NASH-HCC serum samples that cholesterol and fatty acid metabolism regulators, LXR/RXR and FXR/RXR, along with acute phase response signaling, complement system, and clathrin-mediated endocytosis signaling are involved in pathways related to glycoproteins in the development of NASH and HCC [16]. Alpha-2-macroglobulin (A2MG), haptoglobin (HP), apolipoprotein C3 (APOC3), complement factor H (CFAH), serotransferrin (TRFE), vitronectin precursor (VTNC), ceruloplasmin (CERU), and alpha-1 antitrypsin (A1AT) exhibited unidirectional differences in abundance across the three phenotypes. Among the upstream regulators participating in NASH-associated hepatocarcinogenesis, HNF1α, HNF4α, and sterol regulatory element binding factor (SREBF1) were identified as associated with NASH HCC transcription factors. Zhang et al. found that alpha-1-acid glycoprotein (AGP1) glycopeptides had significantly higher glycan branching, sialylation, and fucosylation in samples from NASH and cirrhosis patients compared to controls. This suggests that AGP-1 glycan could potentially serve as a diagnostic biomarker for NASH and associated HCC [29][38]. In addition, Kamada et al. discovered that fucosylated and hyper-sialylated variants of HP are valuable indicators for distinguishing NASH from NAFLD and HCC from control groups [30][39]. HP glycoforms could be therefore applied as markers for the diagnosis of NASH and associated HCC. In addition to various glycosylated protein markers, the APOC3 protein exhibits notably decreased levels in NASH HCC, and can be applied for NASH-HCC diagnosis [31][40]. Moreover, specific glycopeptide moieties located at amino acid position 1424 of A2MG are detectable in the plasma of HCC patients [31][40].2.5. Immune Dysregulation and Activation of Liver Fibrogenesis with the Central Role for TGF-β/SMAD Pathway
In the pathogenesis of NAFLD and NASH, an increase in plasma insulin levels, de novo lipogenesis, TG, and hepatic gluconeogenesis finally resulted in lipotoxicity, damage to hepatocytes, and an influx of immune cells, activation of transforming growth factor β (TGF-β)/SMAD signaling in hepatic stellate cells (HSCs), their differentiation into myofibroblasts, and finally proceeding to progressive fibrosis in the chronic state of NASH [61,62][41][42]. It has been shown that stress-related signaling pathways, like c-Jun N-terminal kinase (JNK) and NF-κB, are activated by pro-inflammatory cytokines, such as TNF-α, IL-1, IL-6, STAT3, and ERK. This activation leads to increased hepatocyte proliferation and ultimately promotes the development of hepatocarcinogenesis [61][41]. HSCs that are not strongly activated generate growth factors and cytokines, including hepatocyte growth factor (HGF), which can help prevent hepatocyte death and the development of HCC [62][42]. However, in the case of chronic liver injury, a shift to tumor-promoting HSC occurs, giving rise to a highly active HSC with myofibroblastic phenotype with increased deposition of type I collagen and other matrix proteins, which promotes cell proliferation and secreting TGF-β and platelet-derived growth factor (PDGF) [63][43]. In addition, TGF-β activation boosts the production of TGF-β within the cell and extends the lifespan of activated HSCs by decreasing apoptosis [64][44]. In addition, integrins, which act as cell surface mechanoreceptors, can interfere with TGF-β1 and PDGF and modulate the proliferation and survival of hepatocytes and HSC [65][45]. The regenerative repair pathways, such as the Hedgehog signaling pathway, are induced during the process of fibrogenesis in NASH and predict the induction of hepatocarcinogenesis [61][41]. Besides the hepatocytes, tumor microenvironment stromal cells, endothelial cells, immune cells, cytokines, and extracellular matrix (ECM) have been suggested to play a key role in the initiation and progression of HCC [66,67][46][47]. In proteomic studies with human NASH-associated biopsies, in addition to alteration proteins associated with inflammation, lipid deposition, insulin resistance, and oxidative stress, significant elevation of numerous ECM and cytoskeleton proteins, with the majority being downstream of TGF-β, including different collagens, fibronectin, vimentin, beta actin-like protein 2, tropomyosin α-4 chain, myosin 9, tubulin α-1C chain, moesin, and others, have been demonstrated [13,68][13][48]. On the contrary, the intermediate filament members CK8 and CK18 were strongly downregulated in human NASH biopsies and HCCs but not in virus-associated HCCs [13,69][13][49]. The CK18 fragments in the blood can indicate NASH and the severity of NAFLD in patients with uncontrolled hepatocyte apoptosis, necrosis, and caspase cleavage. A previous study on a diverse group of patients with biopsy-proven NAFLD highlights the potential clinical value of this test [32][50]. In addition, a large ECM glycoprotein thrombospondin-I (TSP-1) was reported to be a potential target of interest in NAFLD/NASH as it can help to develop anti-fibrotic therapeutics [33][51]. TSP-1 regulates inflammation, cell adhesion, angiogenesis, and several signaling pathways, such as TGF-β1. A comparison of the transcriptomic profiles based on genotype revealed significant differences in the expression of the PPAR pathway and amino acid metabolism pathways in TSP-1 null mice. These findings were further supported by a noticeable decrease in serum lipid levels, indicating activation of the PPAR pathway [33][51]. The influence of gut bacterial dysbiosis and microbial metabolites in the promotion of NASH hepatocarcinogenesis, worsening liver disease through enterohepatic circulation, has been recently reported [70,71][52][53]. It is known that the liver carries out immunological functions and participates in the physiological connection between gut-derived molecules and the systemic circulation, in which liver macrophages, Kupffer cells (KCs) producing pro-inflammatory and pro-fibrogenic cytokines and chemokines, liver resident cells, and monocytes play a very important role [61][41]. M1 macrophages promote inflammation, whereas M2 macrophages exert inhibitory effects and induce tissue repair. Moreover, pro-inflammatory cytokines, including TNF-α, IL-1β, IL-6, IL-12, C-C chemokine ligand 2 (CCL2) and 5 (CCL5) are secreted by M1 macrophages and exhibit increased amounts of nitric oxide species (NOS) and ROS [61][41]. Obesity, whether caused by diet or genetics, can affect the gut microbiota and increase the levels of deoxycholic acid (DCA), a metabolite produced by gut bacteria. This can lead to the activation of senescence-associated secretory phenotype (SASP) in hepatic stellate cells (HSCs), resulting in the secretion of inflammatory and tumor-promoting factors (such as IL-1, 6, and 8) in the liver. This process can contribute to the development of hepatocellular carcinoma (HCC) in both human and mouse models [8]. Recent metabolome research has revealed a reduction in serum levels of indole-3-propionic acid, a potential biomarker and a deamination metabolite of tryptophan maintaining intestinal homeostasis, in NASH-HCC patients [12,34][12][54]. The use of choline-deficient, L-amino acid-defined, high-fat diet (CDAHFD)-fed NASH model mice in animal experiments revealed severe inflammation, fibrosis, impaired mitochondrial respiration, and severe oxidative stress [72][55]. However, de novo lipogenesis (DLG), gluconeogenesis, and antioxidant enzymes in the liver were found to decrease. These changes were associated with a reduction in mitochondrial DNA copy number and complex proteins [73][56]. When exploring NASH biomarkers with gene expression analysis of the whole blood, a marked increase in the inflammation markers such as TNF-α, monocyte chemoattractant protein-1 (MCP1), IL1-β and lipocalin 2 (LCN2) (inflammation), collagen 1a1 (COL1A1) and 3a1 (COL3A1) (fibrosis), and Nrf2 targets NAD(P)H quinone dehydrogenase 1 (NQO1) and heme oxygenase (HO1) was found in the CDAHFD group. On the other hand, CDAHFD induced suppression of stearoyl-coenzyme A desaturase 1 (SCD1), fatty acid synthase (FASN), SREBF-1 (lipogenesis), G6PC and PEPCK (gluconeogenesis), and GPX2, CAT, and SOD3 (antioxidant enzymes) [73][56]. Recent findings in CDAHFD-treated mice suggested that proteins vitronectin (VN), monoacylglycerol acyltransferase 2 (MGAT2), retinoic acid-inducible gene-I (RIG-I)-like receptor (RLR), and formyl peptide receptor 2 (FPR2) may become potential diagnostic biomarkers and/or promising therapeutic opportunities for NAFLD/NASH by modulating the inflammatory reaction and contributing to the development of fibrosis via the activation of HSCs [35,36,37,38][57][58][59][60]. VN, being an EMT component protein, was shown to increase the COL1a2 mRNA expression levels, decrease CD11b and F4/80, macrophage markers, as well as TNF-α and IL-1β inflammatory cytokines and α-SMA, but did not affect the fatty acid synthases expression [35][57]. The regulation of TG absorption and homeostasis is crucially dependent on the highly expressed T2 enzyme in both the human small intestine and liver [36][58]. RLR and FPR2 could be also involved in inflammatory reactions in NASH development [37][59].2.6. The Role of NRIP1 in NAFLD/NASH and Associated HCC
Recent proteome and bioinformatic analyses of NASH human liver biopsies have revealed a fascinating connection between the development of NAFLD and NASH and the activation of the transcription cofactor NRIP1 (nuclear-receptor-interacting protein 1), also known as RIP140 (receptor-interacting protein, 140 kDa) [13]. NRIP1 is a multifunctional transcriptional co-regulator, which plays important physiological roles in the control of a large number of metabolic nuclear receptors and transcription factors, such as the estrogen-related receptor (ESR1), nuclear receptor subfamily 3 group C member 1 (NR3C1) and member 2 (NR3C2), PPARα, PPARβ/δ, PPARγ, liver-X-receptor (LXRα), retinoid-X-receptor alpha (RXRα), apoAI enhancer, Wnt/β-catenin, RAR related orphan receptor A (RORA), and RORC, and is involved in the regulation of the ARNTL/BMAL1, CLOCK, and CRY1 circadian clock genes expression [74,75,76,77,78][61][62][63][64][65]. The NRIP1 gene plays a crucial role in regulating several bodily functions, including energy expenditure, lipid and glucose metabolism, inflammatory response, intestinal homeostasis, ovulation, mammary gland development, behavior, and cognition [74,79,80][61][66][67]. Compared to their wild-type counterparts on an HFD, NRIP1-null mice exhibit remarkable leanness, lack of obesity and hepatic steatosis, improved glucose tolerance, and heightened insulin responsiveness [81][68]. NRIP1 function as a coactivator for LXR appears to have an impact on its ability to regulate FAS expression in the liver [74,81][61][68]. NRIP1 also participates in the regulation of key steps and various oncogenic signaling pathways during cancer initiation and progression, especially in breast, ovary, liver, and colon tumors, and the development of atherosclerosis [79,82,83][66][69][70]. The impact of NRIP1 on HCC has been studied, and findings indicate that decreased levels of NRIP1 (both mRNA and protein) can lead to increased growth and migration of cancer cells in HCC. This is believed to be due to the direct interaction between NRIP1 and β-catenin, leading to repression of β-catenin/T-cell factor/lymphoid enhancer factor (TCF) signaling [78][65]. Furthermore, in Huh7 and HepG2 human liver cancer cells, overexpression of NRIP1 suppressed the malignant potential of liver cancer cells by inhibiting NF-kappaB (NF-kB)-mediated alternative polarization of macrophages [84][71], while the downregulation of miR-140-3-p miRNA, which targets the NRIP1 mRNA, was suggested to influence the hepatocarcinogenesis by stimulating anti-apoptotic signaling [85][72]. In an adrenoleukodystrophy mouse model, the genetic inactivation of NRIP1 prevented mitochondrial depletion and dysfunction, bioenergetic failure, and inflammatory dysregulation. This was achieved through a redox-dependent mechanism driven by very-long-chain fatty acids [86][73]. From these studies, NRIP1 is implicated in the development of NAFLD/NASH due to metabolic and inflammatory dysregulation and is downregulated in HCC, stimulating anti-apoptotic signaling and acting as an enhancer of proliferation, growth, and malignant potential of liver cancer cells. Further deciphering the link between NRIP1, hormone nuclear receptors, LXR/Fas, PPARs, Wnt/β-catenin/T-cell factor (TCF), and its epigenetic regulation could lead to novel strategies against the development of NASH and associated HCC. Furthermore, the NRIP1 in the form of circulating RNA (circRNA) could become a potential biomarker of NAFLD/NASH-HCC, which needs further investigation.References
- Abdelhamed, W.; El-Kassas, M. Hepatocellular carcinoma and hepatitis C virus treatments: The bold and the beautiful. J. Viral Hepat. 2023, 30, 148–159.
- Araujo, A.R.; Rosso, N.; Bedogni, G.; Tiribelli, C.; Bellentani, S. Global epidemiology of non-alcoholic fatty liver disease/non-alcoholic steatohepatitis: What we need in the future. Liver Int. 2018, 38 (Suppl. S1), 47–51.
- Armandi, A.; Schattenberg, J.M. NAFLD and NASH: The Metabolically Diseased Liver. Handb. Exp. Pharmacol. 2022, 274, 253–267.
- Ito, T.; Nguyen, M.H. Perspectives on the Underlying Etiology of HCC and Its Effects on Treatment Outcomes. J. Hepatocell. Carcinoma 2023, 10, 413–428.
- Facciorusso, A. The influence of diabetes in the pathogenesis and the clinical course of hepatocellular carcinoma: Recent findings and new perspectives. Curr. Diabetes Rev. 2013, 9, 382–386.
- Polyzos, S.A.; Kountouras, J.; Mantzoros, C.S. Leptin in nonalcoholic fatty liver disease: A narrative review. Metabolism 2015, 64, 60–78.
- Loo, T.M.; Kamachi, F.; Watanabe, Y.; Yoshimoto, S.; Kanda, H.; Arai, Y.; Nakajima-Takagi, Y.; Iwama, A.; Koga, T.; Sugimoto, Y.; et al. Gut Microbiota Promotes Obesity-Associated Liver Cancer through PGE2-Mediated Suppression of Antitumor Immunity. Cancer Discov. 2017, 7, 522–538.
- Yoshimoto, S.; Loo, T.M.; Atarashi, K.; Kanda, H.; Sato, S.; Oyadomari, S.; Iwakura, Y.; Oshima, K.; Morita, H.; Hattori, M.; et al. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature 2013, 499, 97–101.
- Nakagawa, H.; Umemura, A.; Taniguchi, K.; Font-Burgada, J.; Dhar, D.; Ogata, H.; Zhong, Z.; Valasek, M.A.; Seki, E.; Hidalgo, J.; et al. ER stress cooperates with hypernutrition to trigger TNF-dependent spontaneous HCC development. Cancer Cell 2014, 26, 331–343.
- Vachher, M.; Bansal, S.; Kumar, B.; Yadav, S.; Arora, T.; Wali, N.M.; Burman, A. Contribution of organokines in the development of NAFLD/NASH associated hepatocellular carcinoma. J. Cell Biochem. 2022, 123, 1553–1584.
- Dhamija, E.; Paul, S.B.; Kedia, S. Non-alcoholic fatty liver disease associated with hepatocellular carcinoma: An increasing concern. Indian. J. Med. Res. 2019, 149, 9–17.
- Ahmed, E.A.; El-Derany, M.O.; Anwar, A.M.; Saied, E.M.; Magdeldin, S. Metabolomics and Lipidomics Screening Reveal Reprogrammed Signaling Pathways toward Cancer Development in Non-Alcoholic Steatohepatitis. Int. J. Mol. Sci. 2022, 24, 210.
- Kakehashi, A.; Stefanov, V.E.; Ishii, N.; Okuno, T.; Fujii, H.; Kawai, K.; Kawada, N.; Wanibuchi, H. Proteome Characteristics of Non-Alcoholic Steatohepatitis Liver Tissue and Associated Hepatocellular Carcinomas. Int. J. Mol. Sci. 2017, 18, 434.
- Kakehashi, A.; Suzuki, S.; Ishii, N.; Okuno, T.; Kuwae, Y.; Fujioka, M.; Gi, M.; Stefanov, V.; Wanibuchi, H. Accumulation of 8-hydroxydeoxyguanosine, L-arginine and Glucose Metabolites by Liver Tumor Cells Are the Important Characteristic Features of Metabolic Syndrome and Non-Alcoholic Steatohepatitis-Associated Hepatocarcinogenesis. Int. J. Mol. Sci. 2020, 21, 7746.
- Okuno, T.; Kakehashi, A.; Ishii, N.; Fujioka, M.; Gi, M.; Wanibuchi, H. mTOR Activation in Liver Tumors Is Associated with Metabolic Syndrome and Non-Alcoholic Steatohepatitis in Both Mouse Models and Humans. Cancers 2018, 10, 465.
- Ramachandran, P.; Xu, G.; Huang, H.H.; Rice, R.; Zhou, B.; Lindpaintner, K.; Serie, D. Serum Glycoprotein Markers in Nonalcoholic Steatohepatitis and Hepatocellular Carcinoma. J. Proteome Res. 2022, 21, 1083–1094.
- Jiao, J.; Gonzalez, A.; Stevenson, H.L.; Gagea, M.; Sugimoto, H.; Kalluri, R.; Beretta, L. Depletion of S100A4+ stromal cells does not prevent HCC development but reduces the stem cell-like phenotype of the tumors. Exp. Mol. Med. 2018, 50, e422.
- Wang, Y.; Shi, M.; Fu, H.; Xu, H.; Wei, J.; Wang, T.; Wang, X. Mammalian target of the rapamycin pathway is involved in non-alcoholic fatty liver disease. Mol. Med. Rep. 2010, 3, 909–915.
- Ginsberg, H.N.; Mani, A. Complex regulation of fatty liver disease. Science 2022, 376, 247–248.
- Guri, Y.; Colombi, M.; Dazert, E.; Hindupur, S.K.; Roszik, J.; Moes, S.; Jenoe, P.; Heim, M.H.; Riezman, I.; Riezman, H.; et al. mTORC2 Promotes Tumorigenesis via Lipid Synthesis. Cancer Cell 2017, 32, 807–823.e12.
- Alshehade, S.; Alshawsh, M.A.; Murugaiyah, V.; Asif, M.; Alshehade, O.; Almoustafa, H.; Al Zarzour, R.H. The role of protein kinases as key drivers of metabolic dysfunction-associated fatty liver disease progression: New insights and future directions. Life Sci. 2022, 305, 120732.
- Feng, J.; Qiu, S.; Zhou, S.; Tan, Y.; Bai, Y.; Cao, H.; Guo, J.; Su, Z. mTOR: A Potential New Target in Nonalcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2022, 23, 9196.
- Yin, G.; Liang, H.; Sun, W.; Zhang, S.; Feng, Y.; Liang, P.; Chen, S.; Liu, X.; Pan, W.; Zhang, F. Shuangyu Tiaozhi decoction alleviates non-alcoholic fatty liver disease by improving lipid deposition, insulin resistance, and inflammation in vitro and in vivo. Front. Pharmacol. 2022, 13, 1016745.
- Yoshida, K.; Yokota, K.; Watanabe, K.; Tsuda, H.; Matsumoto, A.; Mizukami, H.; Iwamoto, S. Lack of GPR180 ameliorates hepatic lipid depot via downregulation of mTORC1 signaling. Sci. Rep. 2023, 13, 1843.
- Liu, K.; Qiu, D.; Liang, X.; Huang, Y.; Wang, Y.; Jia, X.; Li, K.; Zhao, J.; Du, C.; Qiu, X.; et al. Lipotoxicity-induced STING1 activation stimulates MTORC1 and restricts hepatic lipophagy. Autophagy 2022, 18, 860–876.
- Wang, G.; Zhang, X.; Zhou, Z.; Song, C.; Jin, W.; Zhang, H.; Wu, W.; Yi, Y.; Cui, H.; Zhang, P.; et al. Sphingosine 1-phosphate receptor 2 promotes the onset and progression of non-alcoholic fatty liver disease-related hepatocellular carcinoma through the PI3K/AKT/mTOR pathway. Discov. Oncol. 2023, 14, 4.
- Masarone, M.; Troisi, J.; Aglitti, A.; Torre, P.; Colucci, A.; Dallio, M.; Federico, A.; Balsano, C.; Persico, M. Untargeted metabolomics as a diagnostic tool in NAFLD: Discrimination of steatosis, steatohepatitis and cirrhosis. Metabolomics 2021, 17, 12.
- Sugiyama, K.; Yu, L.; Nagasue, N. Direct effect of branched-chain amino acids on the growth and metabolism of cultured human hepatocellular carcinoma cells. Nutr. Cancer 1998, 31, 62–68.
- Hagiwara, A.; Nishiyama, M.; Ishizaki, S. Branched-chain amino acids prevent insulin-induced hepatic tumor cell proliferation by inducing apoptosis through mTORC1 and mTORC2-dependent mechanisms. J. Cell Physiol. 2012, 227, 2097–2105.
- Hardwick, J.P.; Osei-Hyiaman, D.; Wiland, H.; Abdelmegeed, M.A.; Song, B.J. PPAR/RXR Regulation of Fatty Acid Metabolism and Fatty Acid omega-Hydroxylase (CYP4) Isozymes: Implications for Prevention of Lipotoxicity in Fatty Liver Disease. PPAR Res. 2009, 2009, 952734.
- Seki, S.; Kitada, T.; Sakaguchi, H. Clinicopathological significance of oxidative cellular damage in non-alcoholic fatty liver diseases. Hepatol. Res. 2005, 33, 132–134.
- Kakehashi, A.; Wei, M.; Fukushima, S.; Wanibuchi, H. Oxidative stress in the carcinogenicity of chemical carcinogens. Cancers 2013, 5, 1332–1354.
- Iwai, S.; Murai, T.; Makino, S.; Min, W.; Morimura, K.; Mori, S.; Hagihara, A.; Seki, S.; Fukushima, S. High sensitivity of fatty liver Shionogi (FLS) mice to diethylnitrosamine hepatocarcinogenesis: Comparison to C3H and C57 mice. Cancer Lett. 2007, 246, 115–121.
- Lee, S.J.; Kang, J.H.; Iqbal, W.; Kwon, O.S. Proteomic analysis of mice fed methionine and choline deficient diet reveals marker proteins associated with steatohepatitis. PLoS ONE 2015, 10, e0120577.
- Ma, Y.; Zhang, G.; Kuang, Z.; Xu, Q.; Ye, T.; Li, X.; Qu, N.; Han, F.; Kan, C.; Sun, X. Empagliflozin activates Sestrin2-mediated AMPK/mTOR pathway and ameliorates lipid accumulation in obesity-related nonalcoholic fatty liver disease. Front. Pharmacol. 2022, 13, 944886.
- Jiao, L.; Wei, J.; Ye, J.; Zhang, C. Prognostic Value of Peroxiredoxin-1 Expression in Patients with Solid Tumors: A Meta-Analysis of Cohort Study. Dis. Markers 2021, 2021, 9508702.
- Lopez-Grueso, M.J.; Lagal, D.J.; Garcia-Jimenez, A.F.; Tarradas, R.M.; Carmona-Hidalgo, B.; Peinado, J.; Requejo-Aguilar, R.; Barcena, J.A.; Padilla, C.A. Knockout of PRDX6 induces mitochondrial dysfunction and cell cycle arrest at G2/M in HepG2 hepatocarcinoma cells. Redox Biol. 2020, 37, 101737.
- Zhang, D.; Huang, J.; Luo, D.; Feng, X.; Liu, Y.; Liu, Y. Glycosylation change of alpha-1-acid glycoprotein as a serum biomarker for hepatocellular carcinoma and cirrhosis. Biomark. Med. 2017, 11, 423–430.
- Kamada, Y.; Ono, M.; Hyogo, H.; Fujii, H.; Sumida, Y.; Mori, K.; Tanaka, S.; Yamada, M.; Akita, M.; Mizutani, K.; et al. A novel noninvasive diagnostic method for nonalcoholic steatohepatitis using two glycobiomarkers. Hepatology 2015, 62, 1433–1443.
- Ji, E.S.; Hwang, H.; Park, G.W.; Lee, J.Y.; Lee, H.K.; Choi, N.Y.; Jeong, H.K.; Kim, K.H.; Kim, J.Y.; Lee, S.; et al. Analysis of fucosylation in liver-secreted N-glycoproteins from human hepatocellular carcinoma plasma using liquid chromatography with tandem mass spectrometry. Anal. Bioanal. Chem. 2016, 408, 7761–7774.
- van Son, K.C.; Verschuren, L.; Hanemaaijer, R.; Reeves, H.; Takkenberg, R.B.; Drenth, J.P.H.; Tushuizen, M.E.; Holleboom, A.G. Non-Parenchymal Cells and the Extracellular Matrix in Hepatocellular Carcinoma in Non-Alcoholic Fatty Liver Disease. Cancers 2023, 15, 1308.
- Filliol, A.; Saito, Y.; Nair, A.; Dapito, D.H.; Yu, L.X.; Ravichandra, A.; Bhattacharjee, S.; Affo, S.; Fujiwara, N.; Su, H.; et al. Opposing roles of hepatic stellate cell subpopulations in hepatocarcinogenesis. Nature 2022, 610, 356–365.
- Tsuchida, T.; Friedman, S.L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 397–411.
- Saile, B.; Matthes, N.; Knittel, T.; Ramadori, G. Transforming growth factor beta and tumor necrosis factor alpha inhibit both apoptosis and proliferation of activated rat hepatic stellate cells. Hepatology 1999, 30, 196–202.
- Rokugawa, T.; Konishi, H.; Ito, M.; Iimori, H.; Nagai, R.; Shimosegawa, E.; Hatazawa, J.; Abe, K. Evaluation of hepatic integrin alphavbeta3 expression in non-alcoholic steatohepatitis (NASH) model mouse by 18F-FPP-RGD2 PET. EJNMMI Res. 2018, 8, 40.
- Loomba, R.; Friedman, S.L.; Shulman, G.I. Mechanisms and disease consequences of nonalcoholic fatty liver disease. Cell 2021, 184, 2537–2564.
- Ogunwobi, O.O.; Harricharran, T.; Huaman, J.; Galuza, A.; Odumuwagun, O.; Tan, Y.; Ma, G.X.; Nguyen, M.T. Mechanisms of hepatocellular carcinoma progression. World J. Gastroenterol. 2019, 25, 2279–2293.
- Du, D.; Liu, C.; Qin, M.; Zhang, X.; Xi, T.; Yuan, S.; Hao, H.; Xiong, J. Metabolic dysregulation and emerging therapeutical targets for hepatocellular carcinoma. Acta Pharm. Sin. B 2022, 12, 558–580.
- Younossi, Z.M.; Wong, G.; Anstee, Q.M.; Henry, L. The Global Burden of Liver Disease. Clin. Gastroenterol. Hepatol. 2023, 21, 978–1991.
- Feldstein, A.E.; Wieckowska, A.; Lopez, A.R.; Liu, Y.C.; Zein, N.N.; McCullough, A.J. Cytokeratin-18 fragment levels as noninvasive biomarkers for nonalcoholic steatohepatitis: A multicenter validation study. Hepatology 2009, 50, 1072–1078.
- Min-DeBartolo, J.; Schlerman, F.; Akare, S.; Wang, J.; McMahon, J.; Zhan, Y.; Syed, J.; He, W.; Zhang, B.; Martinez, R.V. Thrombospondin-I is a critical modulator in non-alcoholic steatohepatitis (NASH). PLoS ONE 2019, 14, e0226854.
- Tian, Y.; Li, Y.; Wang, W.X.; Jiang, W.L.; Fei, J.; Li, C.Y. Novel Strategy for Validating the Existence and Mechanism of the “Gut-Liver Axis” in Vivo by a Hypoxia-Sensitive NIR Fluorescent Probe. Anal. Chem. 2020, 92, 4244–4250.
- Albhaisi, S.A.M.; Bajaj, J.S.; Sanyal, A.J. Role of gut microbiota in liver disease. Am. J. Physiol. Gastrointest. Liver Physiol. 2020, 318, G84–G98.
- Sehgal, P.B. Interleukin-6 at the Host-Tumor Interface: STAT3 in Biomolecular Condensates in Cancer Cells. Cells 2022, 11, 1164.
- Suzuki-Kemuriyama, N.; Abe, A.; Nakane, S.; Uno, K.; Ogawa, S.; Watanabe, A.; Sano, R.; Yuki, M.; Miyajima, K.; Nakae, D. Nonobese mice with nonalcoholic steatohepatitis fed on a choline-deficient, l-amino acid-defined, high-fat diet exhibit alterations in signaling pathways. FEBS Open Bio 2021, 11, 2950–2965.
- Sugasawa, T.; Ono, S.; Yonamine, M.; Fujita, S.I.; Matsumoto, Y.; Aoki, K.; Nakano, T.; Tamai, S.; Yoshida, Y.; Kawakami, Y.; et al. One Week of CDAHFD Induces Steatohepatitis and Mitochondrial Dysfunction with Oxidative Stress in Liver. Int. J. Mol. Sci. 2021, 22, 5851.
- Hayashida, M.; Hashimoto, K.; Ishikawa, T.; Miyamoto, Y. Vitronectin deficiency attenuates hepatic fibrosis in a non-alcoholic steatohepatitis-induced mouse model. Int. J. Exp. Pathol. 2019, 100, 72–82.
- Cheng, D.; Zinker, B.A.; Luo, Y.; Shipkova, P.; De Oliveira, C.H.; Krishna, G.; Brown, E.A.; Boehm, S.L.; Tirucherai, G.S.; Gu, H.; et al. MGAT2 inhibitor decreases liver fibrosis and inflammation in murine NASH models and reduces body weight in human adults with obesity. Cell Metab. 2022, 34, 1732–1748.e5.
- Kawaguchi, S.; Sakuraba, H.; Horiuchi, M.; Ding, J.; Matsumiya, T.; Seya, K.; Iino, C.; Endo, T.; Kikuchi, H.; Yoshida, S.; et al. Hepatic Macrophages Express Melanoma Differentiation-Associated Gene 5 in Nonalcoholic Steatohepatitis. Inflammation 2022, 45, 343–355.
- Lee, C.; Kim, J.; Han, J.; Oh, D.; Kim, M.; Jeong, H.; Kim, T.J.; Kim, S.W.; Kim, J.N.; Seo, Y.S.; et al. Formyl peptide receptor 2 determines sex-specific differences in the progression of nonalcoholic fatty liver disease and steatohepatitis. Nat. Commun. 2022, 13, 578.
- Fritah, A.; Christian, M.; Parker, M.G. The metabolic coregulator RIP140: An update. Am. J. Physiol. Endocrinol. Metab. 2010, 299, E335–E340.
- Harnish, D.C.; Evans, M.J.; Scicchitano, M.S.; Bhat, R.A.; Karathanasis, S.K. Estrogen regulation of the apolipoprotein AI gene promoter through transcription cofactor sharing. J. Biol. Chem. 1998, 273, 9270–9278.
- Jakobsson, T.; Osman, W.; Gustafsson, J.A.; Zilliacus, J.; Warnmark, A. Molecular basis for repression of liver X receptor-mediated gene transcription by receptor-interacting protein 140. Biochem. J. 2007, 405, 31–39.
- Miyata, K.S.; McCaw, S.E.; Meertens, L.M.; Patel, H.V.; Rachubinski, R.A.; Capone, J.P. Receptor-interacting protein 140 interacts with and inhibits transactivation by, peroxisome proliferator-activated receptor alpha and liver-X-receptor α. Mol. Cell Endocrinol. 1998, 146, 69–76.
- Zhang, D.; Wang, Y.; Dai, Y.; Wang, J.; Suo, T.; Pan, H.; Liu, H.; Shen, S. Downregulation of RIP140 in hepatocellular carcinoma promoted the growth and migration of the cancer cells. Tumour Biol. 2015, 36, 2077–2085.
- Lapierre, M.; Docquier, A.; Castet-Nicolas, A.; Gitenay, D.; Jalaguier, S.; Teyssier, C.; Cavailles, V. The emerging role of the transcriptional coregulator RIP140 in solid tumors. Biochim. Biophys. Acta 2015, 1856, 144–150.
- Nautiyal, J.; Christian, M.; Parker, M.G. Distinct functions for RIP140 in development, inflammation, and metabolism. Trends Endocrinol. Metab. 2013, 24, 451–459.
- Leonardsson, G.; Steel, J.H.; Christian, M.; Pocock, V.; Milligan, S.; Bell, J.; So, P.W.; Medina-Gomez, G.; Vidal-Puig, A.; White, R.; et al. Nuclear receptor corepressor RIP140 regulates fat accumulation. Proc. Natl. Acad. Sci. USA 2004, 101, 8437–8442.
- He, Y.; Zhang, L.; Li, Z.; Gao, H.; Yue, Z.; Liu, Z.; Liu, X.; Feng, X.; Liu, P. RIP140 triggers foam-cell formation by repressing ABCA1/G1 expression and cholesterol efflux via liver X receptor. FEBS Lett. 2015, 589, 455–460.
- Yu, X.H.; Xue, X.; Zhu, X.; Li, X. Downregulation of RIP140 in triple-negative breast cancer inhibits the growth and proliferation of cancer cells. Oncol. Lett. 2018, 15, 8784–8788.
- Hu, Y.C.; Yi, Z.J.; Zhou, Y.; Li, P.Z.; Liu, Z.J.; Duan, S.G.; Gong, J.P. Overexpression of RIP140 suppresses the malignant potential of hepatocellular carcinoma by inhibiting NF-κB-mediated alternative polarization of macrophages. Oncol. Rep. 2017, 37, 2971–2979.
- Takata, A.; Otsuka, M.; Kojima, K.; Yoshikawa, T.; Kishikawa, T.; Yoshida, H.; Koike, K. MicroRNA-22 and microRNA-140 suppress NF-κB activity by regulating the expression of NF-κB coactivators. Biochem. Biophys. Res. Commun. 2011, 411, 826–831.
- Ranea-Robles, P.; Galino, J.; Espinosa, L.; Schluter, A.; Ruiz, M.; Calingasan, N.Y.; Villarroya, F.; Naudi, A.; Pamplona, R.; Ferrer, I.; et al. Modulation of mitochondrial and inflammatory homeostasis through RIP140 is neuroprotective in an adrenoleukodystrophy mouse model. Neuropathol. Appl. Neurobiol. 2022, 48, e12747.
More