Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Catherine Yang and Version 1 by Riti Sharan.
Tuberculosis (TB) and Human Immunodeficiency Virus (HIV) co-infection continues to pose a significant healthcare burden. HIV co-infection during TB predisposes the host to the reactivation of latent TB infection (LTBI), worsening disease conditions and mortality. There is a lack of biomarkers of LTBI reactivation and/or immune-related transcriptional signatures to distinguish active TB from LTBI and predict TB reactivation upon HIV co-infection. Characterizing individual cells using next-generation sequencing-based technologies has facilitated novel biological discoveries about infectious diseases, including TB and HIV pathogenesis.
- TB/HIV co-infection
- single cell analysis
- latent TB infection
- biomarkers
1. Introduction
The Tuberculosis (TB) and Human Immunodeficiency Virus (HIV) co-pandemic continues to pose a major healthcare burden in resource-limited countries [1]. TB is caused by Mycobacterium tuberculosis (Mtb), which spreads from person to person through aerosol droplets when the infected person coughs or speaks [2]. According to the Global Tuberculosis Report by the World Health Organization (WHO), an estimated 187,000 people with HIV died of TB in 2021 [3]. People living with HIV (PLHIV) are more likely to develop active TB disease than those without HIV [4,5][4][5]. This is due to several factors, including increased susceptibility to new Mtb infection and, especially, the reactivation of the untreated latent TB infection upon immunosuppression by HIV co-infection [6].
Both pathogens attack the immune subsets needed for protection in the human body: macrophages and CD4+ T cells [7,8][7][8]. However, the mechanisms of this manipulation of the immune responses are not well understood. HIV-1 depletes CD4+ T cells and infects macrophages, leading to virus persistence in reservoirs. Mtb pathogenesis is characterized by phagocytosis of the inhaled bacilli by alveolar macrophages. Upon internalization, Mtb is either cleared, or it proliferates within the macrophages [9]. One of the hallmark features of the immune response to Mtb is the formation of granuloma consisting of macrophages, multinucleated giant cells, Foamy cells, and lymphocytes [10]. Though the primary function of granuloma is to contain the bacilli, in some instances, the bacilli can survive inside these structures for extended periods of time in a dormant state. Under immunosuppressive conditions, such as HIV co-infection, the bacilli escape the granuloma and spread to extrapulmonary organs causing the reactivation of LTBI [6,11,12][6][11][12]. Nonhuman primate (NHP) research has shown that there are direct cytopathic effects of Simian Immunodeficiency Virus (SIV) resulting in chronic immune activation, altered effector T cell phenotypes, and dysregulated T cell homeostasis that causes LTBI reactivation [13]. Studies have also shown that while HIV-1 can replicate in the tuberculous microenvironment [14], Mtb is able to exacerbate HIV-1 pathogenesis through the formation of membranous structures in human macrophages [15]. Concomitant to this, PLHIV with LTBI have been shown to have elevated levels of immune activation and inflammation that impacts the progression of both diseases [16,17][16][17].
Highly effective combinatorial antiretroviral therapy (cART), while effective in reducing viral loads in the periphery and lungs of Mtb/SIV co-infected macaques, fails to reduce the rate of reactivation of LTBI [18,19][18][19]. Despite being on cART, people living with HIV (PLHIV) have increased risk of LTBI reactivation. This is partly due to the inability of cART to significantly reduce the virus-driven chronic immune activation that persists despite undetectable HIV viral loads [20,21][20][21]. This is, in turn, due to a loss of mucosal integrity in the gastrointestinal tract following HIV infection. This leads to the release of microbial products into the circulation, in turn leading to the systemic activation of a wide array of immune cells, such as T, B, NK, plasmacytoid dendritic cells (pDCs), and monocytes [1,22][1][22]. Additionally, the initiation of cART in humans with chronic HIV-1 infection can lead to a paradoxical worsening of TB and manifest as an immune-reconstituted inflammatory syndrome (TB-IRIS) [23].
2. scRNA-Seq—A Tool to Study Host–Pathogen Interactions and Biosignatures in TB
Pulmonary TB, caused by infection with Mtb, is primarily identified as a lung disease wherein the bacilli are able to evade the host immune system in ~5–7% of individuals and cause extensive inflammation and pathology [49][24]. In 90% of the infected individuals, the infection is controlled by the host immune response, and the mycobacterial replication is contained within granulomatous structures [50][25]. This results in LTBI that can either last for the lifetime of the individual or it can reactivate in an immunosuppressed condition, such as HIV co-infection [51][26]. The interaction of the immune cells and complex intracellular bacterial pathogens, such as Mtb, produces multidimensional interactions. Transcriptional profiling at the single-cell level within the dynamic granulomatous structure provides an opportunity to understand the infection as a consolidated process between the host and pathogen (Figure 1). Indeed, heterogeneity is integral to the interactions between intracellular bacteria and host immune cells such as macrophages [52][27]. Additionally, there is evidence that variation in the host and pathogen transcriptional programs causes differential infection outcomes [53,54][28][29]. Further, each granuloma within an individual differs substantially in its composition, inflammatory profile, size, and bacterial ecology [10]. Cellular compositions and cell-to-cell signaling networks have been identified using high-throughput scRNA-seq within TB lung granulomas in the cynomolgus macaque model of TB [55][30]. Granulomas with persistent bacteria were characterized as having TH2 immunity-based signaling among mast, endothelial, fibroblast, and plasma cells. Granulomas demonstrating bacterial control appeared enriched in cytotoxic T cells signaling via TH1, TH17 immunity [55][30]. In a similar approach to studying heterogeneity in the cellular composition of leprosy granulomas, scRNA-seq was utilized to perform Uniform Manifold Approximation and Projection (UMAP) dimensionality reduction and cell clustering [56][31]. This was followed by differential expression analysis to find the cluster markers that defined signature genes. This ultimately resulted in the identification of 66 key antimicrobial genes expressed by T cells, macrophages, keratinocytes, and fibroblasts that have been reported to participate in the immune response to mycobacterial infections [56][31]. By using scRNA-seq in combination with other technologies, both these studies were able to demonstrate the participation of the non-traditional immune cells in a spatial model of mycobacterial infection. Future studies must further elucidate these observations to outline the mechanisms involved in the HIV-1-mediated disruption of granulomas in LTBI.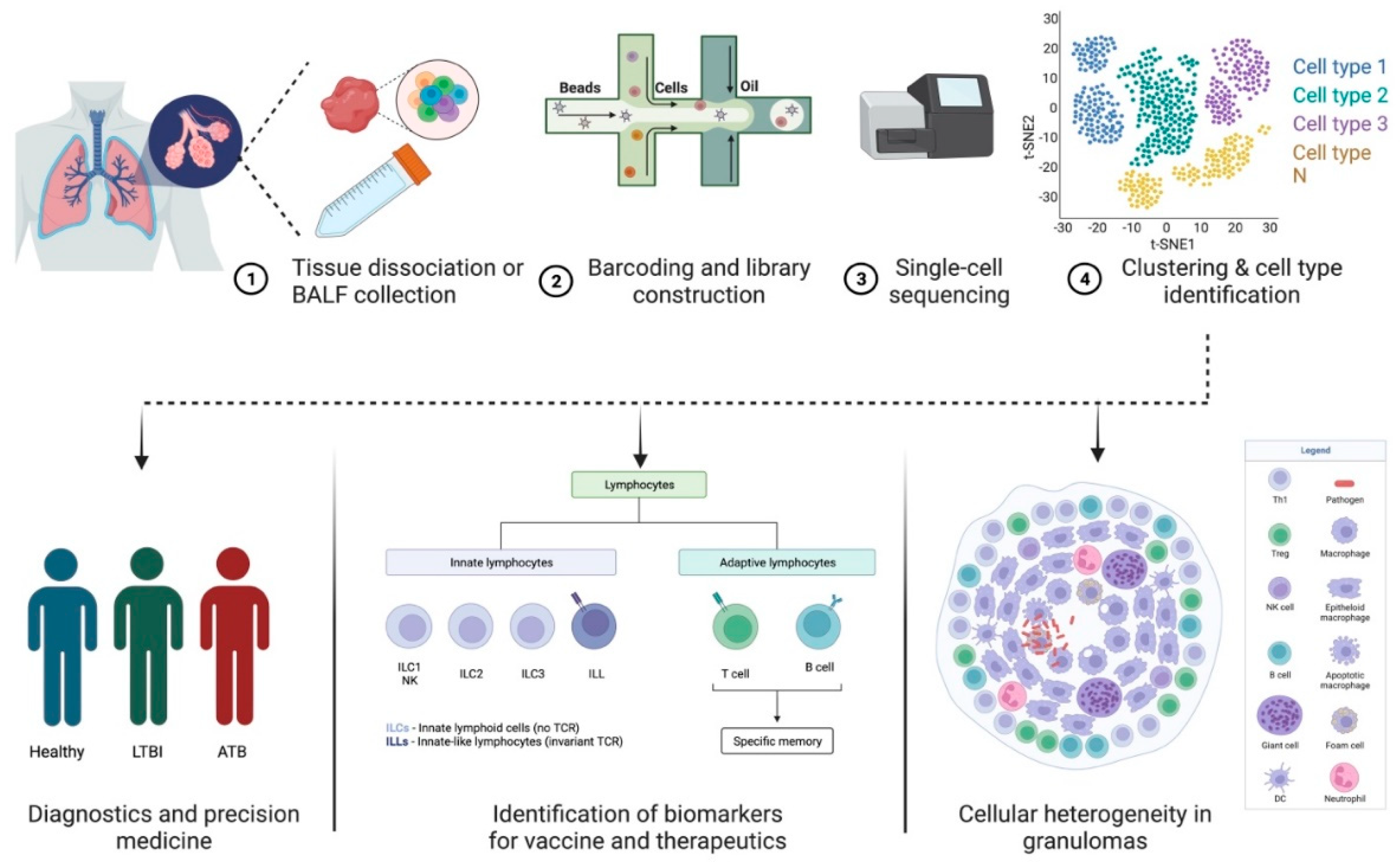
Figure 1. Applications of scRNA-seq in TB pathogenesis and diagnostics. (1) Lung or bronchoalveolar lavage fluid is collected from infected patients. (2) Single-cell suspension prepared from the sample is subjected to (3) barcoded library construction. (4) The cDNA libraries undergo next-generation sequencing that can be analyzed using different visualization tools. Data generated using scRNA-seq techniques can be used to study cellular heterogeneity in granulomas and identify biomarkers for vaccines and therapeutics in diagnostics and precision medicine. The figure was made with help of Biorender.
3. Predicting Disease Progression in TB and HIV Using scRNA-Seq
Infections are dynamic processes in which the outcomes are dictated by the complex interactions of the pathogen and the multiple biological factors of the host. scRNA-seq allows a deeper understanding of the human cellular landscape by studying cellular behavior at a higher resolution. The early diagnosis of LTBI and screening of those at a higher risk of HIV co-infection is imperative for TB prevention programs [105][37]. A simultaneous epitope and transcriptome measurement, using Cellular Indexing of Transcriptomes and Epitopes (CITE-seq), identified a T-cell state associated with TB progression that responded to ex vivo Mtb peptide stimulation [106][38]. The study was based on the fact that TB progression was largely influenced by host memory T cells, which had been shown previously. However, prior studies have failed to comprehensively describe the landscape of the progression-associated memory T cells in TB [107,108][39][40]. Most of the significant differences resided in a rare multimodally defined TH17 subset (C-12). The C-12’s abundance in patients that had recovered from TB indicated either a prior TB disease or susceptibility to TB disease progression. Additionally, mutations in the RAR-related orphan receptor C (RORC), that are highly expressed in C-12, were shown to increase susceptibility to TB [106,109][38][41]. The study provides a resource to enable further studies of the different memory T cell phenotypes that could serve as biomarkers for progression to TB in the setting of HIV co-infection. In a different approach by Ben-Moshe et al., scRNA-seq of human peripheral blood cells infected with Salmonella was used to develop a deconvoluted algorithm to study three cohorts of TB patients at different stages of the disease [110][42]. The trained algorithm predicted the power of the monocyte-infection-induced signature in classifying the LTBI individuals at a higher risk of developing active TB. The study highlights the role of global infection dynamics, such as the monocyte-infection-induced state in Salmonella and TB. The approach can be applied to additional co-morbid pathogens, such as HIV, and at additional time points in order to train the deconvolution further to differentiate the infection-induced states and infectious outcomes. Early interleukin-1 (IL-1) signaling enrichment using scRNA-seq has been implicated as an important pathway in response to Mtb stimulation in human peripheral blood cells [111][43]. The study provided a detailed analysis of the significance of cell-type specificity compared to pathogen-specificity and its role in influencing gene expression in TB. Concomitantly, elevated levels of monocytic IL-1b upon TLR-2 and TLR-4 stimulation have been shown to be associated with reduced odds of TB recurrence in ART-treated patients [112][44]. Innovative single-cell technologies indicate efforts to shift from traditional therapy in TB to precision medicine and host-directed therapeutics. scRNA-seq allows the simultaneous analysis of the host and pathogen transcriptomics on a wide array of samples ranging from primary cell cultures to animal tissue biopsies or blood from the human host. Future studies could focus on utilizing single-cell technology in combination with spatial transcriptomics to understand the impact of gut microbiota, especially in the context of HIV co-infection in humans. HIV acquisition and the clinical outcomes of HIV-1 infection are highly variable among individuals. Genetic factors, socio-economic backgrounds, and complex interactions between the virus and the host determine them. Besides the genetic variation of major host factors, such as human leukocyte antigen receptor (HLA) and HIV co-receptor CCR5, polymorphisms at several loci affect the outcomes of HIV infection as well as the pharmacokinetics of anti-HIV drugs (reviewed in reference [113][45]). Nevertheless, even among the most highly exposed individuals, a fraction remains HIV-negative without any known genetic association [114,115][46][47]. Similarly, important differences in the natural course of HIV infection (such as time from infection to AIDS diagnosis and opportunistic infections or malignancies) were only partially explained by known variables such as genetic polymorphism, age, and co-morbidities [116][48]. Single-cell transcriptome studies, performed in patient-derived cells and in vitro infected primary cells and cell lines, have significantly contributed to understanding the host and virus variability that may alter the overall clinical course of infection (Table 1; Figure 2).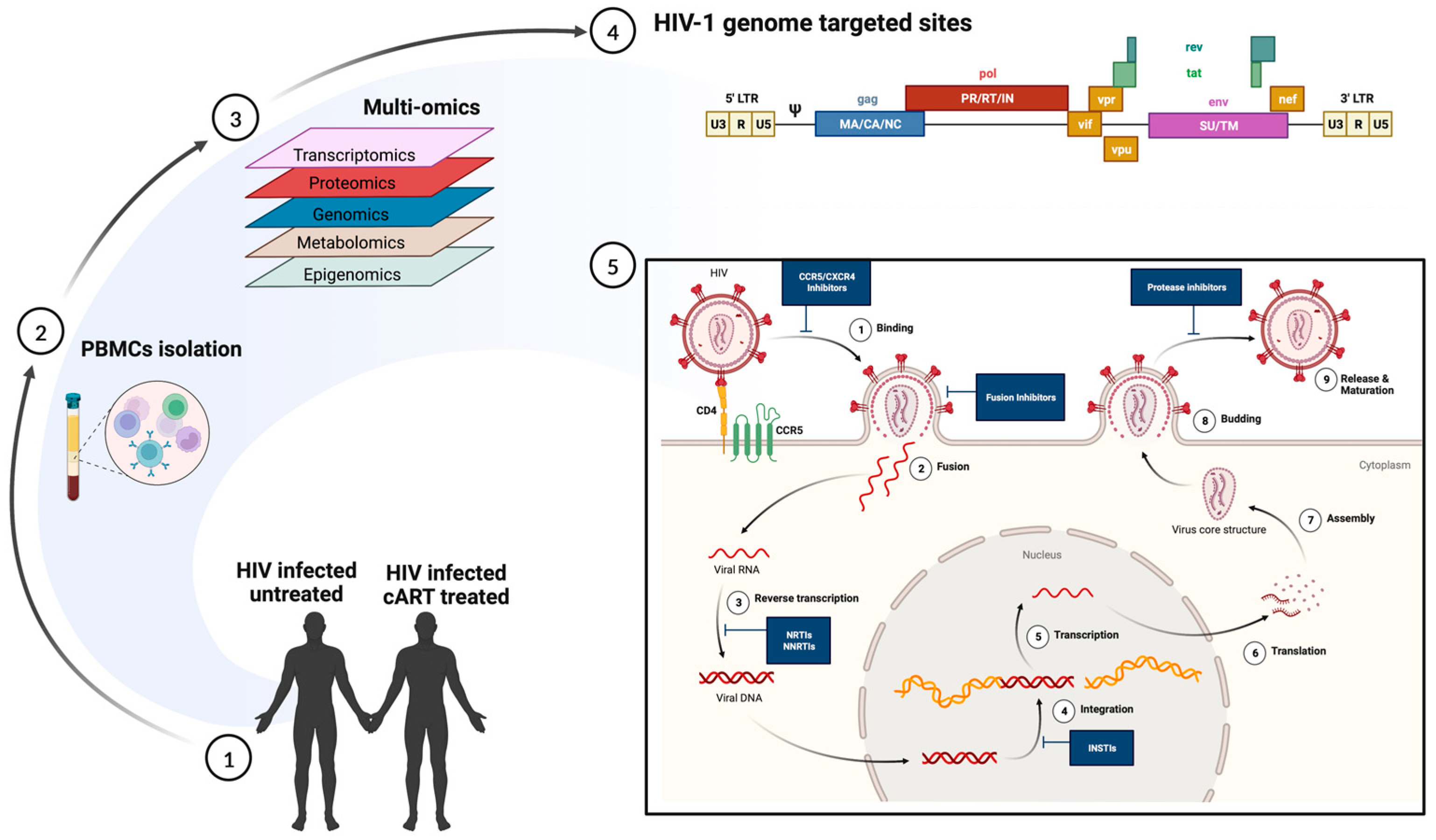
Figure 2. Multi-omics approach to understanding heterogeneity in HIV infection. (1) and (2) Polymorphonuclear cells, collected from HIV-infected untreated and HIV-infected cART-treated individuals, are subjected to multi-omics (3) including transcriptomics, proteomics, genomics, metabolomics, and epigenomics. The data generated can enable (4) an understanding of the transcriptionally active regions in productive infection and (5) help identify sites of therapeutic interventions. The figure was made with help of Biorender.
Table 1.
HIV acquisition and the clinical outcomes of HIV-1 infection.
| Study | Technique | Observation | Ref. |
|---|---|---|---|
| Defining HIV-permissive cells | scRNA-seq | TCR stimulation mainly contributes to transcriptional heterogeneity in CD4+ T cells, which regulates HIV transcription. | [120][53] |
| Immune responses during hyperacute HIV infection | scRNA-seq on peripheral blood mononuclear cells from four untreated individuals before and longitudinally during acute infection |
Immune cell responses to HIV infection within the first weeks of infection, such as proliferating natural killer cells, which potentially may be associated with viral control | [30][55] |
| Restoration of T cell function after ART | scRNA-seq on peripheral T cells on chronic HIV-infected treatment-naïve or ART-treated patients | Significant loss of naïve T cells, prolonged inflammation, and increased response to interferon-α in treatment-naïve patients, partially restored by ART. Granulosyn expressing CD4+ and CD8+ Effector cell clusters correlated with poor immune restoration |
[123][57] |
| Immune Exhaustion in Chronic HIV Infection | scRNA-seq on peripheral blood mononuclear cells from HIV patients and healthy subjects | An inhibitory receptor KLRG1 was identified in an HIV-1 specific exhausted CD8+ T cell population expressing KLRG1, TIGIT, and Tbetdim Eomeshi markers | [124][58] |
| Immune Reconstitution failure in (HAART)-treated HIV patients | scRNA-seq and scATAC-seq analysis of peripheral blood mononuclear cells (PBMCs) derived from immune non-responder [127][61] and responder (IR) | Low expression of mucosal-associated invariant T (MAIT) cells in INRs, which exhibited transcriptional profiles associated with impaired mitochondrial function and apoptosis signaling | [125][59] |
| Cellular transcriptome changes induced by active HIV replication in macrophages | An in vitro system to model HIV-1 infection of macrophages and single-cell RNA sequencing (scRNA-seq) to compare the transcriptomes of uninfected cells, cells harboring pre-integration complexes (PIC), and those containing integrated provirus and making late HIV proteins | NFkB- and AP-1-promoted transcription characterize PIC cell transcriptomes, while E2F family transcription products distinguish transcriptomes of cells transcribing from provirus | [121][54] |
| Differential virus reactivation potential | Primary CD4+ T-cell model expression HIV green fluorescent protein (GFP); scRNA seq | Global transcriptomic profiles of cells with reactivated HIV showed higher cellular activation and metabolic activity | [28][62] |
| Characterize latent cells reactivated by a single round of stimulation | Latent cell capture; LURE. Purified CD4+ T cells from peripheral blood of ART-treated patients activated by LRS and sorted based on the expression of HIV-env protein and HIV-gag mRNA | CD4+ T cells harboring proviruses with identical Env sequences also showed identical TCRs. Reservoir cells arise by clonal expansion, and the reservoir is maintained by balanced cell division and cell death. Reactivated latent cells express a distinct transcriptional program that suppresses HIV-1 transcription, which helps them survive | [78][63] |
| Characterize reactivated cells within 24 h of latency reversal | Sortseq; stimulated peripheral blood CD4+ T cells from ART-treated, virally suppressed patients | HIV+ cells enriched in TH1 phenotype upregulate cellular factors that support HIV transcription and promote cellular survival. HIV promoter drives high aberrant host gene transcription downstream of the integration site | [79][64] |
| Transcriptome of quiescent reservoir memory CD4+ T cells harboring intact HIV provirus | Quiescent reservoir memory CD4+ T cells enriched using their unique TCR as a molecule, identified using scRNA-seq | HIV–proviral integration and latency did not induce a specific transcriptional program | [85][65] |
| Characterize HIV reservoir cells after suppressive ART-therapy | ECCITE-seq, which captures surface protein expression, cellular transcriptome, HIV-1 RNA, and TCR sequence within the same single cells in longitudinally archived paired samples during actual viremia and one-year post-ART | HIV revised in heterogenous granzyme B+ Th1 effector memory CD4+ T cells with robust antigen response, proliferation potential, and long-term clonal stability | [80][66] |
| Isolate reservoir cells from HIV patients treated with long-term ART | FIND-seq, simultaneous capture of polyadenylated RNA and HIV DNA from single cells, and scRNA-seq without latency reversal | HIV-DNA+ memory CD4+ T cells inhibited death receptor, necroptosis, and antiproliferative signaling but showed higher expression of known negative regulators of HIV transcription, thus favoring long-term survival of the cells and HIV silencing | [88][67] |
| The transcriptome of HIV-infected and HIV-exposed peripheral blood monocytes | scRNA seq | Monocytes provide varying degrees of permissive cellular environment for HIV infection, and ART has a detrimental effect on monocyte function | [94][68] |
| scRNA profiling of HIV-infected cells in CNS | Single nucleus RNA-seq of archived brain tissue | HIV-induced interferon response modulates host chromatin conformation and HIV integration sites in microglia | [95][69] |
4. Implications of scRNA-Seq in Animal Models of TB and HIV
The nonhuman primate (NHP) model remains the most accurate animal model that recapitulates the entire spectrum of TB as seen in humans [129,130][71][72]. Rhesus macaque (Macaca mulatta) is the most widely used model amongst all the NHP species. This is due to their social adaptability and genetic diversity, which makes them adept at investigating intra-specific variation [131][73]. The model is highly tractable and can be utilized to study latent TB and LTBI reactivation and can be co-infected with Simian Immunodeficiency Virus (SIV) [13,18,19,132][13][18][19][74]. Cataloging the diverse cellular architecture of the primate lung in LTBI and active TB is critical in understanding human disease progression. The multi-omics approach provides an atlas that serves as an open resource for identifying novel targets for disease interventions. scRNA-seq on dematricized single-cells from lungs of active-pulmonary-TB-infected rhesus macaques identified an influx of plasmacytoid dendritic cells (pDCs), the interferon (IFN)-responsive macrophage population, and activated T-cell responses [133][75]. On the other hand, LTBI lungs were characterized by a CD27+ natural killer cell subset. The study outlined the presence of unique immune populations associated with either TB control or dissemination, which could be targeted for therapeutics or vaccines in the future. Several studies have also attempted to generate an NHP cell atlas using scRNA-seq to provide a catalog of features that can be used to study human disease [134,135][76][77]. These studies have mapped the receptor and co-receptors for viruses causing human infectious diseases, then intersected the data with human genetic disease orthologues for translational associations. A novel approach combined bacterial fitness fluorescent reporter strains with scRNA-seq to dissect the functional heterogeneity of Mtb-infected alveolar and interstitial macrophages in a mouse model [136][78]. The approach showed that, while the local lung macrophage population was epigenetically constrained in response to the infection, an inter-species comparison confirmed the conserved nature of the macrophage subsets between mice and humans. Such a multimodal scRNA-seq approach has important implications for understanding the regulation of host cells before and during TB/HIV co-infection. Akter et al. employed scRNA-seq on Mtb-infected murine lungs to identify the enrichment of a type I Interferon (IFN) signature in the lymphocytes and a higher expression of Ly6A on the surface of activated lymphocytes [60][36]. The results are in concordance with the human TB signatures [137][79] and identify Ly6A as a critical marker of T-cell activation in TB. The dynamic changes in T-cell subsets during TB infection, measured using scRNA-seq, revealed a novel signature for exhausted CD4+ (H1FX, ZFP36L2, VIM, PPP1R15A) and CD8+ T cells (ITM2C) [138][80]. T-cell exhaustion has important implications in immune deficiency, exacerbation of acute TB, and CD8+ T cell function. The identification of different gene signatures and pathways associated with exhausted T-cell subsets can potentially facilitate a comprehensive understanding of the role of these subsets in Mtb pathogenesis in the setting of HIV co-infection. Similar to TB, the diverse and complex pathogenesis of HIV-1 remains a challenge in developing efficacious vaccines and cure strategies. The absence of treatment results in a highly variable disease progression with considerable heterogeneity in the phenotype of infected immune cells and their functions [139][81]. The heterogeneous nature of the HIV-infected cells also makes it difficult to target and treat the latent reservoirs [28][62]. The single-cell approach allows the study of the diverse mechanisms associated with HIV latency, including transcriptional repression and post-transcriptional blocks [140][82]. The effects of chronic HIV infection, including the persistence of viral reservoirs and neuropathogenesis, can be mimicked using Simian Immunodeficiency Virus (SIV) in an infected rhesus macaque model. The transcriptome of brain myeloid cells in cART-treated versus untreated macaques was examined using scRNA-seq. The results demonstrated that the virus induced changes in the brain microglia and that cART restored the homeostatic state of microglia like the uninfected control [141][83]. The findings provide a critical perspective on the role of microglia/macrophages as key targets of the SIV infection of the central nervous system. This finding has direct implications in elucidating the underlying mechanisms of alteration of the clinical signs and delays in the diagnosis of neurotuberculosis, the most devastating extrapulmonary form of TB in the setting of HIV infection. The differences in the neonatal versus adult responses to the HIV-1 envelope were demonstrated using a macaque model [142][84]. Single-cell transcriptomic analyses showed that neonatal macaques had a decreased immunosuppression transcriptome signature compared to adult macaques following HIV envelope immunization. Additionally, the neonates had a high transcriptome profile of BCL2 in T cells and decreased IL10RA transcripts in T, B, and NK cells post-HIV envelope vaccination compared to adult macaques. These findings are especially critical to advance our understanding of the transcriptomic changes in neonates who are exposed to HIV and have TB. Though the NHP model of SIV has unequivocally illustrated the mechanisms of HIV pathogenesis, there are significant genetic and biological differences between lentiviruses and some HIV-specific interventions that cannot be tested in nonhuman hosts. Humanized mice represent an attractive alternative animal model that recapitulates key aspects of human HIV biology and pathogenesis [143,144][85][86]. Single-cell transcriptome analysis of human CD45+CD3-CD19- cells isolated from the spleens of humanized mice demonstrated a significant upregulation of the genes involved in type I IFN inflammatory pathways in the innate immune subsets [145][87]. In another study by Aso et al., the characterization of HIV-1-producing cells in a humanized mouse model using scRNA-seq demonstrated the heterogeneity of HIV-1-infected cells, including CXCL13high cells that provide clues for the development of an HIV-1 cure [146][88]. Humanized mice also support dual infections of Mtb and HIV in the lung, CNS, and other organs [147,148,149][89][90][91]. Co-infection in humanized mice reproduces several important aspects of microbial synergy, such as greater viral and bacterial burden and exacerbated inflammation. Applying single-cell RNA sequencing technology in this model would further enable the profiling of the complex virus, bacterial, and host dynamics during infection (Figure 3). It is a powerful tool that helps improve our understanding of the intercellular communication networks and the host–bacteria/virus interactions in immunologically relevant animal models.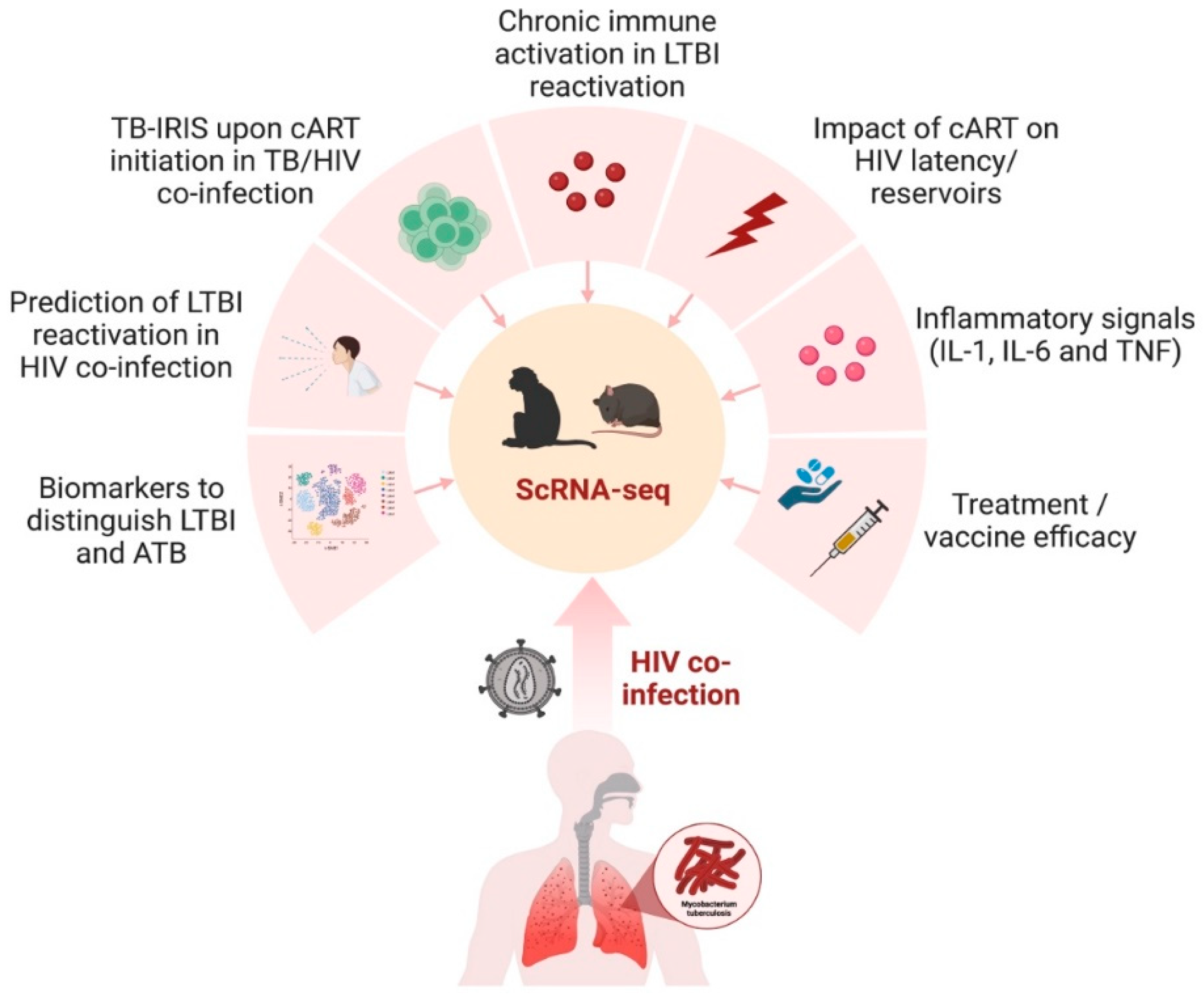
Figure 3. scRNA-seq in preclinical animal models to study TB/HIV co-infection. The nonhuman primate and humanized mouse model can be utilized to gain transcriptomic insights into several aspects of TB and HIV co-infection in humans, including the impact of treatment, diagnostics, inflammation, and immune activation. The figure was made with help of Biorender.
References
- Sharan, R.; Bucşan, A.N.; Ganatra, S.; Paiardini, M.; Mohan, M.; Mehra, S.; Khader, S.A.; Kaushal, D. Chronic Immune Activation in TB/HIV Co-infection. Trends Microbiol. 2020, 28, 619–632.
- Jones-López, E.C.; Namugga, O.; Mumbowa, F.; Ssebidandi, M.; Mbabazi, O.; Moine, S.; Mboowa, G.; Fox, M.P.; Reilly, N.; Ayakaka, I.; et al. Cough aerosols of Mycobacterium tuberculosis predict new infection: A household contact study. Am. J. Respir. Crit. Care Med. 2013, 187, 1007–1015.
- World Health Organization. Global Tuberculosis Report 2022. 2022. Available online: https://www.who.int/teams/global-tuberculosis-programme/tb-reports/global-tuberculosis-report-2022 (accessed on 29 May 2023).
- Adhikari, N.; Bhattarai, R.B.; Basnet, R.; Joshi, L.R.; Tinkari, B.S.; Thapa, A.; Joshi, B. Prevalence and associated risk factors for tuberculosis among people living with HIV in Nepal. PLoS ONE 2022, 17, e0262720.
- Runels, T.; Ragan, E.J.; Ventura, A.S.; Winter, M.R.; White, L.F.; Horsburgh, C.R.; Samet, J.H.; Saitz, R.; Jacobson, K.R. Testing and treatment for latent tuberculosis infection in people living with HIV and substance dependence: A prospective cohort study. BMJ Open 2022, 12, e058751.
- Shea, K.M.; Kammerer, J.S.; Winston, C.A.; Navin, T.R.; Horsburgh, C.R., Jr. Estimated rate of reactivation of latent tuberculosis infection in the United States, overall and by population subgroup. Am. J. Epidemiol. 2014, 179, 216–225.
- Bruchfeld, J.; Correia-Neves, M.; Källenius, G. Tuberculosis and HIV Coinfection. Cold Spring Harb. Perspect. Med. 2015, 5, a017871.
- Lavalett, L.; Rodriguez, H.; Ortega, H.; Sadee, W.; Schlesinger, L.S.; Barrera, L.F. Alveolar macrophages from tuberculosis patients display an altered inflammatory gene expression profile. Tuberculosis 2017, 107, 156–167.
- Guo, Q.; Zhong, Y.; Wang, Z.; Cao, T.; Zhang, M.; Zhang, P.; Huang, W.; Bi, J.; Yuan, Y.; Ou, M.; et al. Single-cell transcriptomic landscape identifies the expansion of peripheral blood monocytes as an indicator of HIV-1-TB co-infection. Cell Insight 2022, 1, 100005.
- McCaffrey, E.F.; Donato, M.; Keren, L.; Chen, Z.; Delmastro, A.; Fitzpatrick, M.B.; Gupta, S.; Greenwald, N.F.; Baranski, A.; Graf, W.; et al. The immunoregulatory landscape of human tuberculosis granulomas. Nat. Immunol. 2022, 23, 318–329.
- Tilahun, M.; Shibabaw, A.; Kiflie, A.; Bewket, G.; Abate, E.; Gelaw, B. Latent tuberculosis infection and associated risk factors among people living with HIV and apparently healthy blood donors at the University of Gondar referral hospital, Northwest Ethiopia. BMC Res. Notes 2019, 12, 515.
- Hoerter, A.; Arnett, E.; Schlesinger, L.S.; Pienaar, E. Systems biology approaches to investigate the role of granulomas in TB-HIV coinfection. Front. Immunol. 2022, 13, 1014515.
- Bucşan, A.N.; Chatterjee, A.; Singh, D.K.; Foreman, T.W.; Lee, T.H.; Threeton, B.; Kirkpatrick, M.G.; Ahmed, M.; Golden, N.; Alvarez, X.; et al. Mechanisms of reactivation of latent tuberculosis infection due to SIV coinfection. J. Clin. Investig. 2019, 129, 5254–5260.
- Bell, L.C.K.; Noursadeghi, M. Pathogenesis of HIV-1 and Mycobacterium tuberculosis co-infection. Nat. Rev. Microbiol. 2018, 16, 80–90.
- Souriant, S.; Balboa, L.; Dupont, M.; Pingris, K.; Kviatcovsky, D.; Cougoule, C.; Lastrucci, C.; Bah, A.; Gasser, R.; Poincloux, R.; et al. Tuberculosis Exacerbates HIV-1 Infection through IL-10/STAT3-Dependent Tunneling Nanotube Formation in Macrophages. Cell Rep. 2019, 26, 3586–3599.
- Wyndham-Thomas, C.; Corbière, V.; Selis, E.; Payen, M.C.; Goffard, J.C.; Van Vooren, J.P.; Mascart, F.; Dirix, V. Immune Activation by Mycobacterium tuberculosis in HIV-Infected and -Uninfected Subjects. J. Acquir. Immune Defic. Syndr. 2017, 74, 103–111.
- LaVergne, S.; Umlauf, A.; McCutchan, A.; Heaton, R.; Benson, C.; Kumarasamy, N.; Bharti, A.R. Impact of Latent Tuberculosis Infection on Neurocognitive Functioning and Inflammation in HIV-Infected and Uninfected South Indians. J. Acquir. Immune Defic. Syndr. 2020, 84, 430–436.
- Sharan, R.; Ganatra, S.R.; Bucsan, A.N.; Cole, J.; Singh, D.K.; Alvarez, X.; Gough, M.; Alvarez, C.; Blakley, A.; Ferdin, J.; et al. Antiretroviral therapy timing impacts latent tuberculosis infection reactivation in a Mycobacterium tuberculosis/SIV coinfection model. J. Clin. Investig. 2022, 132.
- Ganatra, S.R.; Bucşan, A.N.; Alvarez, X.; Kumar, S.; Chatterjee, A.; Quezada, M.; Fish, A.; Singh, D.K.; Singh, B.; Sharan, R.; et al. Antiretroviral therapy does not reduce tuberculosis reactivation in a tuberculosis-HIV coinfection model. J. Clin. Investig. 2020, 130, 5171–5179.
- Teklu, A.M.; Nega, A.; Mamuye, A.T.; Sitotaw, Y.; Kassa, D.; Mesfin, G.; Belayihun, B.; Medhin, G.; Yirdaw, K. Factors Associated with Mortality of TB/HIV Co-infected Patients in Ethiopia. Ethiop. J. Health Sci. 2017, 27, 29–38.
- Zicari, S.; Sessa, L.; Cotugno, N.; Ruggiero, A.; Morrocchi, E.; Concato, C.; Rocca, S.; Zangari, P.; Manno, E.C.; Palma, P. Immune Activation, Inflammation, and Non-AIDS Co-Morbidities in HIV-Infected Patients under Long-Term ART. Viruses 2019, 11, 200.
- Paiardini, M.; Müller-Trutwin, M. HIV-associated chronic immune activation. Immunol. Rev. 2013, 254, 78–101.
- Wilkinson, K.A.; Meintjes, G.; Seldon, R.; Goliath, R.; Wilkinson, R.J. Immunological characterisation of an unmasking TB-IRIS case. S. Afr. Med. J. 2012, 102, 512–517.
- Chandra, P.; Grigsby, S.J.; Philips, J.A. Immune evasion and provocation by Mycobacterium tuberculosis. Nat. Rev. Microbiol. 2022, 20, 750–766.
- Gideon, H.P.; Flynn, J.L. Latent tuberculosis: What the host “sees”? Immunol. Res. 2011, 50, 202–212.
- Wong, N.S.; Leung, C.C.; Chan, K.C.W.; Chan, W.K.; Lin, A.W.C.; Lee, S.S. A longitudinal study on latent TB infection screening and its association with TB incidence in HIV patients. Sci. Rep. 2019, 9, 10093.
- Liao, S.Y.; Atif, S.M.; Mould, K.; Konigsberg, I.R.; Fu, R.; Davidson, E.; Li, L.; Fontenot, A.P.; Maier, L.A.; Yang, I.V. Single-cell RNA sequencing identifies macrophage transcriptional heterogeneities in granulomatous diseases. Eur. Respir. J. 2021, 57, 2003794.
- Avraham, R.; Haseley, N.; Brown, D.; Penaranda, C.; Jijon, H.B.; Trombetta, J.J.; Satija, R.; Shalek, A.K.; Xavier, R.J.; Regev, A.; et al. Pathogen Cell-to-Cell Variability Drives Heterogeneity in Host Immune Responses. Cell 2015, 162, 1309–1321.
- Avraham, R.; Hung, D.T. A perspective on single cell behavior during infection. Gut Microbes 2016, 7, 518–525.
- Gideon, H.P.; Hughes, T.K.; Tzouanas, C.N.; Wadsworth, M.H., 2nd; Tu, A.A.; Gierahn, T.M.; Peters, J.M.; Hopkins, F.F.; Wei, J.R.; Kummerlowe, C.; et al. Multimodal profiling of lung granulomas in macaques reveals cellular correlates of tuberculosis control. Immunity 2022, 55, 827–846.
- Ma, F.; Hughes, T.K.; Teles, R.M.B.; Andrade, P.R.; de Andrade Silva, B.J.; Plazyo, O.; Tsoi, L.C.; Do, T.; Wadsworth, M.H., 2nd; Oulee, A.; et al. The cellular architecture of the antimicrobial response network in human leprosy granulomas. Nat. Immunol. 2021, 22, 839–850.
- Cai, Y.; Dai, Y.; Wang, Y.; Yang, Q.; Guo, J.; Wei, C.; Chen, W.; Huang, H.; Zhu, J.; Zhang, C.; et al. Single-cell transcriptomics of blood reveals a natural killer cell subset depletion in tuberculosis. EBioMedicine 2020, 53, 102686.
- Xu, Y.; Tan, Y.; Zhang, X.; Cheng, M.; Hu, J.; Liu, J.; Chen, X.; Zhu, J. Comprehensive identification of immuno-related transcriptional signature for active pulmonary tuberculosis by integrated analysis of array and single cell RNA-seq. J. Infect. 2022, 85, 534–544.
- Kulkarni, V.; Queiroz, A.T.L.; Sangle, S.; Kagal, A.; Salvi, S.; Gupta, A.; Ellner, J.; Kadam, D.; Rolla, V.C.; Andrade, B.B.; et al. A Two-Gene Signature for Tuberculosis Diagnosis in Persons with Advanced HIV. Front. Immunol. 2021, 12, 631165.
- Hillman, H.; Khan, N.; Singhania, A.; Dubelko, P.; Soldevila, F.; Tippalagama, R.; DeSilva, A.D.; Gunasena, B.; Perera, J.; Scriba, T.J.; et al. Single-cell profiling reveals distinct subsets of CD14+ monocytes drive blood immune signatures of active tuberculosis. Front. Immunol. 2022, 13, 1087010.
- Akter, S.; Chauhan, K.S.; Dunlap, M.D.; Choreño-Parra, J.A.; Lu, L.; Esaulova, E.; Zúñiga, J.; Artyomov, M.N.; Kaushal, D.; Khader, S.A. Mycobacterium tuberculosis infection drives a type I IFN signature in lung lymphocytes. Cell Rep. 2022, 39, 110983.
- Estévez, O.; Anibarro, L.; Garet, E.; Pallares, Á.; Barcia, L.; Calviño, L.; Maueia, C.; Mussá, T.; Fdez-Riverola, F.; Glez-Peña, D.; et al. An RNA-seq Based Machine Learning Approach Identifies Latent Tuberculosis Patients with an Active Tuberculosis Profile. Front. Immunol. 2020, 11, 1470.
- Nathan, A.; Beynor, J.I.; Baglaenko, Y.; Suliman, S.; Ishigaki, K.; Asgari, S.; Huang, C.C.; Luo, Y.; Zhang, Z.; Lopez, K.; et al. Multimodally profiling memory T cells from a tuberculosis cohort identifies cell state associations with demographics, environment and disease. Nat. Immunol. 2021, 22, 781–793.
- Boisson-Dupuis, S.; Bustamante, J.; El-Baghdadi, J.; Camcioglu, Y.; Parvaneh, N.; El Azbaoui, S.; Agader, A.; Hassani, A.; El Hafidi, N.; Mrani, N.A.; et al. Inherited and acquired immunodeficiencies underlying tuberculosis in childhood. Immunol. Rev. 2015, 264, 103–120.
- Scriba, T.J.; Kalsdorf, B.; Abrahams, D.A.; Isaacs, F.; Hofmeister, J.; Black, G.; Hassan, H.Y.; Wilkinson, R.J.; Walzl, G.; Gelderbloem, S.J.; et al. Distinct, specific IL-17- and IL-22-producing CD4+ T cell subsets contribute to the human anti-mycobacterial immune response. J. Immunol. 2008, 180, 1962–1970.
- Okada, S.; Markle, J.G.; Deenick, E.K.; Mele, F.; Averbuch, D.; Lagos, M.; Alzahrani, M.; Al-Muhsen, S.; Halwani, R.; Ma, C.S.; et al. Impairment of immunity to Candida and Mycobacterium in humans with bi-allelic RORC mutations. Science 2015, 349, 606–613.
- Bossel Ben-Moshe, N.; Hen-Avivi, S.; Levitin, N.; Yehezkel, D.; Oosting, M.; Joosten, L.A.B.; Netea, M.G.; Avraham, R. Predicting bacterial infection outcomes using single cell RNA-sequencing analysis of human immune cells. Nat. Commun. 2019, 10, 3266.
- Oelen, R.; de Vries, D.H.; Brugge, H.; Gordon, M.G.; Vochteloo, M.; Ye, C.J.; Westra, H.J.; Franke, L.; van der Wijst, M.G.P. Single-cell RNA-sequencing of peripheral blood mononuclear cells reveals widespread, context-specific gene expression regulation upon pathogenic exposure. Nat. Commun. 2022, 13, 3267.
- Thobakgale, C.; Naidoo, K.; McKinnon, L.R.; Werner, L.; Samsunder, N.; Karim, S.A.; Ndung’u, T.; Altfeld, M.; Naidoo, K. Interleukin 1-Beta (IL-1β) Production by Innate Cells Following TLR Stimulation Correlates with TB Recurrence in ART-Treated HIV-Infected Patients. J. Acquir. Immune Defic. Syndr. 2017, 74, 213–220.
- McLaren, P.J.; Fellay, J. HIV-1 and human genetic variation. Nat. Rev. Genet. 2021, 22, 645–657.
- Horton, R.E.; McLaren, P.J.; Fowke, K.; Kimani, J.; Ball, T.B. Cohorts for the study of HIV-1-exposed but uninfected individuals: Benefits and limitations. J. Infect. Dis. 2010, 202 (Suppl. S3), S377–S381.
- Kulkarni, P.S.; Butera, S.T.; Duerr, A.C. Resistance to HIV-1 infection: Lessons learned from studies of highly exposed persistently seronegative (HEPS) individuals. AIDS Rev. 2003, 5, 87–103.
- Sabin, C.A.; Lundgren, J.D. The natural history of HIV infection. Curr. Opin. HIV AIDS 2013, 8, 311–317.
- Pollara, J.; Khanal, S.; Edwards, R.W.; Hora, B.; Ferrari, G.; Haynes, B.F.; Bradley, T. Single-cell analysis of immune cell transcriptome during HIV-1 infection and therapy. BMC Immunol. 2022, 23, 48.
- Ciuffi, A.; Bleiber, G.; Munoz, M.; Martinez, R.; Loeuillet, C.; Rehr, M.; Fischer, M.; Gunthard, H.F.; Oxenius, A.; Meylan, P.; et al. Entry and transcription as key determinants of differences in CD4 T-cell permissiveness to human immunodeficiency virus type 1 infection. J. Virol. 2004, 78, 10747–10754.
- Mohammadi, P.; Desfarges, S.; Bartha, I.; Joos, B.; Zangger, N.; Munoz, M.; Gunthard, H.F.; Beerenwinkel, N.; Telenti, A.; Ciuffi, A. 24 hours in the life of HIV-1 in a T cell line. PLoS Pathog. 2013, 9, e1003161.
- Rausell, A.; Munoz, M.; Martinez, R.; Roger, T.; Telenti, A.; Ciuffi, A. Innate immune defects in HIV permissive cell lines. Retrovirology 2016, 13, 43.
- Rato, S.; Rausell, A.; Munoz, M.; Telenti, A.; Ciuffi, A. Single-cell analysis identifies cellular markers of the HIV permissive cell. PLoS Pathog. 2017, 13, e1006678.
- Lim, A.L.; Moos, P.; Pond, C.D.; Larson, E.C.; Martins, L.J.; Szaniawski, M.A.; Planelles, V.; Barrows, L.R. HIV-1 provirus transcription and translation in macrophages differs from pre-integrated cDNA complexes and requires E2F transcriptional programs. Virulence 2022, 13, 386–413.
- Kazer, S.W.; Aicher, T.P.; Muema, D.M.; Carroll, S.L.; Ordovas-Montanes, J.; Miao, V.N.; Tu, A.A.; Ziegler, C.G.K.; Nyquist, S.K.; Wong, E.B.; et al. Integrated single-cell analysis of multicellular immune dynamics during hyperacute HIV-1 infection. Nat. Med. 2020, 26, 511–518.
- Mahnke, Y.D.; Fletez-Brant, K.; Sereti, I.; Roederer, M. Reconstitution of Peripheral T Cells by Tissue-Derived CCR4+ Central Memory Cells Following HIV-1 Antiretroviral Therapy. Pathog. Immun. 2016, 1, 260–290.
- Wang, X.M.; Zhang, J.Y.; Xing, X.; Huang, H.H.; Xia, P.; Dai, X.P.; Hu, W.; Zhang, C.; Song, J.W.; Fan, X.; et al. Global transcriptomic characterization of T cells in individuals with chronic HIV-1 infection. Cell Discov. 2022, 8, 29.
- Wang, S.; Zhang, Q.; Hui, H.; Agrawal, K.; Karris, M.A.Y.; Rana, T.M. An atlas of immune cell exhaustion in HIV-infected individuals revealed by single-cell transcriptomics. Emerg. Microbes Infect. 2020, 9, 2333–2347.
- Li, H.; Tang, Y.; Wang, Y.; Li, Y.; Yang, Y.; Liao, K.; Song, F.; Deng, S.; Chen, Y. Single-cell sequencing resolves the landscape of immune cells and regulatory mechanisms in HIV-infected immune non-responders. Cell Death Dis. 2022, 13, 849.
- Niu, M.; Morsey, B.; Lamberty, B.G.; Emanuel, K.; Yu, F.; Leon-Rivera, R.; Berman, J.W.; Gaskill, P.J.; Matt, S.M.; Ciborowski, P.S.; et al. Methamphetamine Increases the Proportion of SIV-Infected Microglia/Macrophages, Alters Metabolic Pathways, and Elevates Cell Death Pathways: A Single-Cell Analysis. Viruses 2020, 12, 1297.
- Tiwari, S.; van Tonder, A.J.; Vilchèze, C.; Mendes, V.; Thomas, S.E.; Malek, A.; Chen, B.; Chen, M.; Kim, J.; Blundell, T.L.; et al. Arginine-deprivation-induced oxidative damage sterilizes Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA 2018, 115, 9779–9784.
- Golumbeanu, M.; Cristinelli, S.; Rato, S.; Munoz, M.; Cavassini, M.; Beerenwinkel, N.; Ciuffi, A. Single-Cell RNA-Seq Reveals Transcriptional Heterogeneity in Latent and Reactivated HIV-Infected Cells. Cell Rep. 2018, 23, 942–950.
- Cohn, L.B.; da Silva, I.T.; Valieris, R.; Huang, A.S.; Lorenzi, J.C.C.; Cohen, Y.Z.; Pai, J.A.; Butler, A.L.; Caskey, M.; Jankovic, M.; et al. Clonal CD4+ T cells in the HIV-1 latent reservoir display a distinct gene profile upon reactivation. Nat. Med. 2018, 24, 604–609.
- Liu, R.; Yeh, Y.J.; Varabyou, A.; Collora, J.A.; Sherrill-Mix, S.; Talbot, C.C., Jr.; Mehta, S.; Albrecht, K.; Hao, H.; Zhang, H.; et al. Single-cell transcriptional landscapes reveal HIV-1-driven aberrant host gene transcription as a potential therapeutic target. Sci. Transl. Med. 2020, 12, eaaz0802.
- Weymar, G.H.J.; Bar-On, Y.; Oliveira, T.Y.; Gaebler, C.; Ramos, V.; Hartweger, H.; Breton, G.; Caskey, M.; Cohn, L.B.; Jankovic, M.; et al. Distinct gene expression by expanded clones of quiescent memory CD4+ T cells harboring intact latent HIV-1 proviruses. Cell Rep. 2022, 40, 111311.
- Collora, J.A.; Liu, R.; Pinto-Santini, D.; Ravindra, N.; Ganoza, C.; Lama, J.R.; Alfaro, R.; Chiarella, J.; Spudich, S.; Mounzer, K.; et al. Single-cell multiomics reveals persistence of HIV-1 in expanded cytotoxic T cell clones. Immunity 2022, 55, 1013–1031.e7.
- Clark, I.C.; Mudvari, P.; Thaploo, S.; Smith, S.; Abu-Laban, M.; Hamouda, M.; Theberge, M.; Shah, S.; Ko, S.H.; Perez, L.; et al. HIV silencing and cell survival signatures in infected T cell reservoirs. Nature 2023, 614, 318–325.
- Leon-Rivera, R.; Morsey, B.; Niu, M.; Fox, H.S.; Berman, J.W. Interactions of Monocytes, HIV, and ART Identified by an Innovative scRNAseq Pipeline: Pathways to Reservoirs and HIV-Associated Comorbidities. mBio 2020, 11.
- Plaza-Jennings, A.L.; Valada, A.; O’Shea, C.; Iskhakova, M.; Hu, B.; Javidfar, B.; Ben Hutta, G.; Lambert, T.Y.; Murray, J.; Kassim, B.; et al. HIV integration in the human brain is linked to microglial activation and 3D genome remodeling. Mol. Cell 2022, 82, 4647–4663.e8.
- Farhadian, S.F.; Mehta, S.S.; Zografou, C.; Robertson, K.; Price, R.W.; Pappalardo, J.; Chiarella, J.; Hafler, D.A.; Spudich, S.S. Single-cell RNA sequencing reveals microglia-like cells in cerebrospinal fluid during virologically suppressed HIV. JCI Insight 2018, 3, e121718.
- Foreman, T.W.; Mehra, S.; Lackner, A.A.; Kaushal, D. Translational Research in the Nonhuman Primate Model of Tuberculosis. ILAR J. 2017, 58, 151–159.
- Scanga, C.A.; Flynn, J.L. Modeling tuberculosis in nonhuman primates. Cold Spring Harb. Perspect. Med. 2014, 4, a018564.
- Cooper, E.B.; Brent, L.J.N.; Snyder-Mackler, N.; Singh, M.; Sengupta, A.; Khatiwada, S.; Malaivijitnond, S.; Qi Hai, Z.; Higham, J.P. The rhesus macaque as a success story of the Anthropocene. Elife 2022, 11, e78169.
- Sharan, R.; Ganatra, S.R.; Singh, D.K.; Cole, J.; Foreman, T.W.; Thippeshappa, R.; Peloquin, C.A.; Shivanna, V.; Gonzalez, O.; Day, C.L.; et al. Isoniazid and rifapentine treatment effectively reduces persistent M. tuberculosis infection in macaque lungs. J. Clin. Investig. 2022, 132, e121718.
- Esaulova, E.; Das, S.; Singh, D.K.; Choreño-Parra, J.A.; Swain, A.; Arthur, L.; Rangel-Moreno, J.; Ahmed, M.; Singh, B.; Gupta, A.; et al. The immune landscape in tuberculosis reveals populations linked to disease and latency. Cell Host Microbe 2021, 29, 165–178.
- Han, L.; Wei, X.; Liu, C.; Volpe, G.; Zhuang, Z.; Zou, X.; Wang, Z.; Pan, T.; Yuan, Y.; Zhang, X.; et al. Cell transcriptomic atlas of the non-human primate Macaca fascicularis. Nature 2022, 604, 723–731.
- Qu, J.; Yang, F.; Zhu, T.; Wang, Y.; Fang, W.; Ding, Y.; Zhao, X.; Qi, X.; Xie, Q.; Chen, M.; et al. A reference single-cell regulomic and transcriptomic map of cynomolgus monkeys. Nat. Commun. 2022, 13, 4069.
- Pisu, D.; Huang, L.; Narang, V.; Theriault, M.; Lê-Bury, G.; Lee, B.; Lakudzala, A.E.; Mzinza, D.T.; Mhango, D.V.; Mitini-Nkhoma, S.C.; et al. Single cell analysis of M. tuberculosis phenotype and macrophage lineages in the infected lung. J. Exp. Med. 2021, 218, e20210615.
- Berry, M.P.; Graham, C.M.; McNab, F.W.; Xu, Z.; Bloch, S.A.; Oni, T.; Wilkinson, K.A.; Banchereau, R.; Skinner, J.; Wilkinson, R.J.; et al. An interferon-inducible neutrophil-driven blood transcriptional signature in human tuberculosis. Nature 2010, 466, 973–977.
- Pan, J.; Zhang, X.; Xu, J.; Chang, Z.; Xin, Z.; Wang, G. Landscape of Exhausted T Cells in Tuberculosis Revealed by Single-Cell Sequencing. Microbiol. Spectr. 2023, 11, e02839-22.
- Sannier, G.; Dubé, M.; Kaufmann, D.E. Single-Cell Technologies Applied to HIV-1 Research: Reaching Maturity. Front. Microbiol. 2020, 11, 297.
- Mbonye, U.; Karn, J. The Molecular Basis for Human Immunodeficiency Virus Latency. Annu. Rev. Virol. 2017, 4, 261–285.
- Trease, A.J.; Niu, M.; Morsey, B.; Guda, C.; Byrareddy, S.N.; Buch, S.; Fox, H.S. Antiretroviral therapy restores the homeostatic state of microglia in SIV-infected rhesus macaques. J. Leukoc. Biol. 2022, 112, 969–981.
- Han, Q.; Bradley, T.; Williams, W.B.; Cain, D.W.; Montefiori, D.C.; Saunders, K.O.; Parks, R.J.; Edwards, R.W.; Ferrari, G.; Mueller, O.; et al. Neonatal Rhesus Macaques Have Distinct Immune Cell Transcriptional Profiles following HIV Envelope Immunization. Cell Rep. 2020, 30, 1553–1569.
- Marsden, M.D. Benefits and limitations of humanized mice in HIV persistence studies. Retrovirology 2020, 17, 7.
- Victor Garcia, J. Humanized mice for HIV and AIDS research. Curr. Opin. Virol. 2016, 19, 56–64.
- Cheng, L.; Yu, H.; Wrobel, J.A.; Li, G.; Liu, P.; Hu, Z.; Xu, X.N.; Su, L. Identification of pathogenic TRAIL-expressing innate immune cells during HIV-1 infection in humanized mice by scRNA-Seq. JCI Insight 2020, 5, e135344.
- Aso, H.; Nagaoka, S.; Kawakami, E.; Ito, J.; Islam, S.; Tan, B.J.Y.; Nakaoka, S.; Ashizaki, K.; Shiroguchi, K.; Suzuki, Y.; et al. Multiomics Investigation Revealing the Characteristics of HIV-1-Infected Cells In Vivo. Cell Rep. 2020, 32, 107887.
- Nusbaum, R.J.; Calderon, V.E.; Huante, M.B.; Sutjita, P.; Vijayakumar, S.; Lancaster, K.L.; Hunter, R.L.; Actor, J.K.; Cirillo, J.D.; Aronson, J.; et al. Pulmonary Tuberculosis in Humanized Mice Infected with HIV-1. Sci. Rep. 2016, 6, 21522.
- Huante, M.B.; Saito, T.B.; Nusbaum, R.J.; Naqvi, K.F.; Chauhan, S.; Hunter, R.L.; Actor, J.K.; Rudra, J.S.; Endsley, M.A.; Lisinicchia, J.G.; et al. Small Animal Model of Post-chemotherapy Tuberculosis Relapse in the Setting of HIV Co-infection. Front. Cell. Infect. Microbiol. 2020, 10, 150.
- Endsley, J.J.; Huante, M.B.; Naqvi, K.F.; Gelman, B.B.; Endsley, M.A. Advancing our understanding of HIV co-infections and neurological disease using the humanized mouse. Retrovirology 2021, 18, 14.
More