1. Cancer-Associated Fibroblasts (CAFs)
Fibroblasts stem from a mesenchymal origin and are considered to be the most abundant cell type present in connective tissue (i.e., the supportive tissue of an organ)
[1][2][69,70]. Through their secretion of collagen proteins, elastins, adhesive proteins (e.g., laminin and fibronectin), and ground substance (e.g., glycosaminoglycans), they maintain integrity of the extracellular matrix (ECM), thereby providing structural support to tissues and organs
[3][71]. Under normal homeostatic conditions, fibroblasts remain quiescent as spindle-shaped single cells embedded within the ECM in the interstitial space, however, in response to tissue damage/injury they become reversibly “activated” to promote tissue repair during the wound healing process
[4][72]. Once “activated” these fibroblasts display enhanced expression of α-smooth muscle actin (αSMA) and vimentin, and, together with increased ECM production and cytoskeleton remodeling, become stellate in shape (i.e., star-like) and gain contractile properties
[5][6], at which point they are identified as being mesenchymal stem cell-like with characteristic signs of smooth muscle that has granted them the name of myofibroblasts.
In many instances, tumors are analogous to a wound as the expansion of new cells and chronic inflammation results in a continuous state of tissue injury, and the chronic activation of a wound healing/fibrotic response, and as such tumors are often referred to as wounds that do not heal
[6][7][8][73,74,75]. Within the TME, fibroblasts that are “activated” by tumor cells are termed cancer-associated fibroblasts (CAFs), which typically constitute the most abundant stromal cell type present within the tumor microenvironment of solid tumors, including those of the colon, breast, and pancreas
[9][76]. In pancreatic cancer for example, the tumor mass typically comprises 60–70% stromal CAFs, enhanced collagen, and other ECM components
[10][77]. The intercellular communication between CAFs and tumor cells occurs via multiple ways, by direct cell–cell contact, by the transfer of secreted molecules, and by secreted extracellular vesicles that include EVs. It has been recently shown that the secretion of EVs is an important way for CAFs to influence the behavior of cancer cells (and vice versa)
[11][12][78,79].
Understanding the role that EVs play in the communication between surrounding stromal cells and epithelial cancer cells, and vice versa, is critical for elucidating the influence of the tumor microenvironment during cancer cell progression and/or metastasis. The inherent mechanisms involved in the “activation” of CAFs within the tumor microenvironment appear to be related to EMT-induced differentiation of both resident stromal cells and recruited bone marrow-derived stem cells, which result in the formation of myofibroblasts. The initiation of CAF “activation” is predominantly driven by transforming growth factor beta 1 (TGF-β1) and fibroblast growth factor-2 (FGF-2)
[13][80]. Interestingly, it has been recently shown that EV-derived TGF-β1 supplied by cancer cells is the only essential requirement for the differentiation of fibroblasts into CAFs
[14][81], supporting the role of EVs for maintenance and modification of the tumor microenvironment. Similarly, ovarian and breast cancer-derived EVs have been shown to induce the conversion of adipose-derived mesenchymal stem cells into myofibroblast-like cells
[15][16][82,83]. Within the TME, CAFs are predominantly responsible for the production of essential extracellular matrix proteins (such as fibronectin and collagens) and proteases
[17][84]. It has been shown that increased production of these proteins leads to the progressive stiffening of the extracellular matrix, thereby facilitating tumor progression, vascularization, and metastasis
[18][85]. The EV release of bioactive molecules, such as platelet-derived growth factor (PDGF), hepatocyte growth factor (HGF), interleukin-6 (IL6), proteases, and miRNAs
[19][86] by CAFs into the extracellular matrix further supports their importance in cell–cell communication within the tumor microenvironment. In breast cancer for example, a study by Luga et al. showed that EVs released from surrounding stromal cells can promote breast cancer cell motility and metastasis through the mobilization of Wnt11-induced planar cell polarity
[20][87]. Indeed, Luga and colleagues demonstrated that EVs released from cancer-associated fibroblasts (CAFs) promote the increased motility and metastatic capability of breast cancer cells, which is dependent on the interaction of breast cancer cell-produced Wnt11 and CAF-derived CD81
[20][87]. Another study on lung cancer cells also showed that microvesicles containing the extracellular matrix MMPs inducer (EMMPRIN) can stimulate the expression of matrix metalloproteinases (MMPs) in CAFs, which results in enhanced tumor metastization
[21][88]. Other studies, performed on prostate cancer cell-derived EVs, which contain significantly increased levels of IL6, TGF-β, MMPs, carbonic anhydrase IX, and tumor necrosis factor 1α (TNF-1α) reported the induction of a stem cell-like phenotype thus enhancing metastasis under hypoxic conditions
[22][23][24][89,90,91].
2. Tumor-Associated Macrophages (TAMs)
Macrophages are a key population of innate immune cells responsible for executing a broad spectrum of functions ranging from the modulation of tissue homeostasis to the defense against pathogens and the facilitation of wound healing
[24][91]. Macrophages that have infiltrated into the microenvironment of solid tumors are termed tumor-associated macrophages (TAMs) and form a critical component of the TME. Many of the TAMS located within the TME originate from bone marrow monocyte precursors and are recruited into the TME as a result of the tumor-derived chemoattractants that are continuously present within tumors. TAMs affect tumor growth, angiogenesis, metastasis, and chemoresistance. Within tumors, most TAMs gather at the leading edge and within hypoxic avascular areas
[25][92], while a few also align along the abluminal side (i.e., away from the lumen) of the vessels
[26][93]. They are recruited and activated by various signals in the TME and can have dramatic impacts on tumor progression and metastasis. TAMs have been demonstrated to perform a diverse range of immune regulatory functions and tumor progression, including that of promoting cancer cell proliferation and invasive capacity. In fact, tumor-elicited inflammation promotes tumor growth via the presence of the TAM-derived inflammatory cytokines interleukin (IL)-23 and IL-17
[27][94]. Moreover, an increase in TAM-derived IL-6 has been shown to contribute to STAT3 signaling induced hepatocellular carcinoma development and progression
[28][95].
Macrophages can display different and even opposing phenotypes, depending on the microenvironment which they are embedded in. Once activated, macrophages are often classified as having either an M1 (classical-activated macrophages) or M2 (alternative-activated macrophages) phenotype
[29][96]. M1 macrophages typically promote an inflammatory response against invading pathogens and tumor cells, whereas M2 macrophages tend to exert an immune-suppressive phenotype, which favors tissue repair and tumor progression. Each polarized macrophage type displays distinct expression profiles of genes, cytokines, and cell-surface markers
[30][97]. Among those factors, colony-stimulating factor 1 (CSF-1) and C-C motif ligand 2 (CCL2) are the two most well-documented macrophage recruiters and M2-stimulating factors. CSF-1 is a potent determinant factor of macrophage polarization, as CSF-1 overexpression is often observed at the invasive edge of various tumors and correlates with a significant increase in metastasis
[31][98].
Many studies have attempted to elucidate the crosstalk that exists between tumor and immune cells within the TME. Studies have shown that tumor-derived EVs play a vital role in the conversion of monocyte-derived macrophages into regulatory macrophages and in the mediation of cancer-related inflammation and tumor development
[32][33][99,100] through the transfer of their cargos to recipient cells within the TME
[34][35][101,102]. These cargos include proteins, nucleic acids, and lipids. Several studies have shown that depletion of EVs can disrupt the communication between tumor cells and TAMs, which reverses some of the harmful effects that EVs exert during tumor progression, restoring chemotherapeutic drug sensitivity
[36][37][38][103,104,105].
In recent years, several studies have shown that EV miRNAs can play a crucial role in tumor progression, through their ability to regulate angiogenesis and facilitate metastasis by interfering with normal cellular programs of recipient cells
[39][40][106,107]. Tumor-derived EV miRNAs have been shown to polarize recipient macrophages by targeting several signaling pathways, which can positively or negatively impact tumor progression
[41][42][65,108]. It has been shown that tumor-derived EV miRNAs can promote cancer metastasis by regulating the crosstalk between cancer cells and TAMs, also providing a therapeutic strategy for cancer therapy. For example, colorectal-derived EVs carrying miR-203 are incorporated into monocytes
[43][109], while EVs carrying miR-145
[44][110] and exosome-like vesicles containing miR-934
[42][108] are taken up by macrophages, which leads to their polarization into the M2 phenotype. M2 polarization induced by EVs derived from oral squamous cell carcinoma harboring miR-29a-3p has been shown to target (SOCS)1/STAT6 signaling, directly promoting tumor growth
[45][111]. Whereas, hypoxia in ovarian cancer induces the production of EVs enriched in miR-940, miR-21-3p, miR-125b-5p, miR-181d-5p, and miR-222-3p stimulates macrophage M2 phenotype polarization and enhanced tumor growth
[46][47][112,113]. Similarly, hypoxia-induced miR-301a-3p-enrichment in lung cancer-derived exosome-like vesicles results in a HIF1a/2a-dependent polarization of TAMs to the M2 phenotype, facilitating enhanced cell invasion, migration, and epithelial–mesenchymal transition (EMT) during lung metastasis
[48][114].
3. Tumor Endothelial Cells (TECs)
Angiogenesis, also known as new blood vessel formation, is essential for tumor progression and metastasis. The onset of angiogenesis occurs at any stage of tumor progression and depends on the type of tumor and its microenvironment. Endothelial cells, in the majority of solid tumors, are located within the inner layer of blood vessels and compared to normal endothelial cells have an altered morphology and molecular phenotype. Tumor blood vessels are characteristically unorganized, where they are often thin, fragile, and defective in barrier function resulting in leakiness of tumor blood vessels, whereas normal vasculature shows a hierarchal branching pattern of arteries, veins, and capillaries
[49][115]. The unorganized nature of tumor blood vessels means that within specific focal regions they lack endothelial cells or basement membrane
[50][116], which results in their exhibited chaotic blood flow (often termed leaky/hemorrhagic)
[51][117]. Additionally, the high interstitial fluid pressure that is present within solid tumors causes blood vessels to collapse and further impedes blood flow. As such, hypoxic regions within the tumor tissue develop, despite the high level of vascularization
[52][118]. During tumor formation and progression epithelial cancer cells actively secrete several pro-angiogenic factors which result in the excessive formation of abnormal blood vessels. Several studies have highlighted the differences between normal endothelial cells and tumor endothelial cells (TECs)
[53][119]. In particular, the release of vascular endothelial growth factor (VEGF) in addition to other growth factors belonging to the ephrin and angiopoietin families from TECs is essential in promoting the formation of tumor blood vessels
[54][120]. Hida et al. demonstrated that when compared to normal endothelial cells, TECs display several abnormalities
[55][56][121,122] including differences in their responsiveness to epidermal growth factor (EGF)
[57][123], adrenomedullin
[58][124], and VEGF
[59][125]. Ultimately, the differences in their response to these growth factors are important in the proangiogenic phenotype of TECs
[56][122]. In particular, VEGF has been shown to stimulate cell migration and enhances survival of TECs in an autocrine manner.
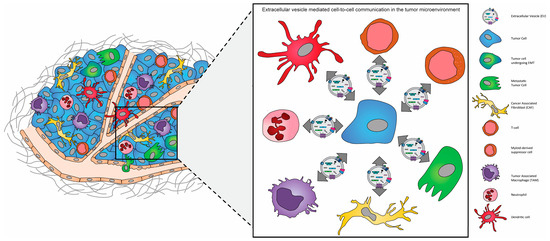
Several studies have demonstrated that like CAFs and TAMs, the infiltration and metabolic switch of endothelial cells to TECs within the tumor microenvironment supports and enhances angiogenesis during tumor growth and morphologically abnormal tumor vasculature promotes tumor cell intravasation during metastasis (
Figure 12). VEGF–VEGF receptor signaling loosens the tight junctions that interconnect adjacent endothelial cells which renders blood vessels permeable to leakage. Additionally, high interstitial fluid pressures coupled with the immature structure of tumor blood vessels enhances the ease by which tumor cells permeate through tumor blood vessels
[60][126]. Several researchers have demonstrated that TECs release specific growth factors, called angiocrine factors
[61][127] into the TEM which convert indolent tumor cells to more aggressive cells displaying greater tumorigenicity, extranodal invasion, and chemoresistance
[62][63][128,129]. Cao et al. showed that fibroblast growth factor 4 (FGF4), produced by B-cell lymphoma cells, activates fibroblast growth factor receptor 1 (FGFR1) in neighboring endothelial cells resulting in the upregulation of the Notch ligand Jagged1. In turn, Jagged1 on endothelial cells reciprocally induces Notch2–Hey1 signaling in lymphoma cells
[62][128]. Mao and colleagues demonstrated that exosome-like vesicles derived from esophageal squamous cell carcinoma (ESCC) cells are mediators of intracellular communication between epithelial cancer cells and vascular endothelial cells within the TME. Specifically, they showed that hypoxic ESCC-derived EVs resulted in increased proliferation, improved capillary-like structure formation, and increased invasive ability of human umbilical endothelial cells (HUVEC), concluding that hypoxic EVs derived from cancer cells alter TECs within the TME, enhancing tumor angiogenesis
[64][130]. To date, many studies have demonstrated that the miRNA, mRNA, and other non-coding RNA cargos of EVs released from tumor cells are responsible for the metabolic reprogramming of stromal cells, including TECs. For example, Chen et al. showed that exosome-like vesicles isolated from the serum of colorectal cancer (CRC) with metastases contain circTUBCGP4 which leads to miR-146b-3p inhibition in HUVEC cells, leading to Akt signaling pathway activation, which results in enhanced cell migration and angiogenic tube formation
[65][131]. Biagoni et al. showed that urokinase plasminogen activator surface receptor (uPAR) containing exosome-like vesicles released from melanoma cells led to an increase in pro-angiogenesis of both human microvascular endothelial cells (HMVECs) and endothelial colony-forming cells (ECFCs), which they demonstrated was as a result of tumor exosome-like vesicle-mediated induction of vascular endothelial cadherin (VE-Cadherin), uPAR, and EGFR protein expression in endothelial cells
[66][132]. In colorectal cancer cell-derived exosome-like vesicles, miR-25-3p has been shown to promote angiogenesis and the disruption of vein endothelial cell tight junctions within distant sites, including the lung and liver, helping to establish the pre-metastatic niche
[67][133].
Figure 12. The “ecosystem” of the tumor microenvironment (TME). The tumor microenvironment of solid tumors is composed of extracellular matrix (ECM) components and a multitude of different stromal cells, including macrophages, dendritic cells, neutrophils, and myeloid-derived suppressor cells, adipocytes, fibroblasts, and endothelial cells, in addition to both non-cancerous and cancerous epithelial cells.
4. Tumor-Infiltrating Lymphocytes (TILs)
Tumor-infiltrating lymphocytes (TILs) are defined as all lymphatic cell populations that invade into solid tumors. They consist primarily of cytotoxic CD8+ T cells and CD4+ helper T cells
[68][134] in addition to smaller proportions of natural killer (NK) and B cells
[69][135]. In all solid tumors TILs play distinct roles in modulating the TME, and for decades their role in tumor progression had been widely debated. In fact, the infiltration of immune cells into the TME and their role in cancer immunosurveillance is one of the hallmarks of cancer
[70][136]. Although TILs have been shown to serve as somewhat of a double-edged sword as their infiltration into tumors can promote the initial establishment of a TME which is more susceptible to enhanced tumor progression
[71][137], they can also attack tumor cells and in that way serve as potent tumor suppressors
[72][138]. During the initial stages of tumor development, the infiltration of TILs and their prolonged interaction with surrounding tumor cells primes the hosts immune system against tumor cell elimination and in that way the tumor immunosurveillance results in the promotion of tumor growth
[73][139]. However, in the long run the constant infiltration of TILs into the TME results in their exhaustion, characterized by sustained expression of inhibitory receptors distinct from functional effector and memory T cells, ultimately resulting in their failure to arrest tumor progression
[72][74][138,140]. In fact, in a variety of tumor types (i.e., breast, colon, lung, and ovarian) the infiltration of immune cells into the TME has been shown to provide value as a predictive prognostic marker
[75][76][77][141,142,143]. The treatment of metastatic cancer remains challenging, and in fact the study by Haj-Shomaly et al., 2022, suggests that paclitaxel chemotherapy, while effective in some cancer instances, may also promote tumor metastasis in the lung through its ability to rapidly induce ECM remodeling mediated by CD8+ T cells expressing lysyl oxidase (LOX), a potent ECM remodeling enzyme
[78][144]. As such, in recent years tumor immunotherapy has become an attractive and effective treatment strategy for many solid tumor types. Several studies highlight the benefits of external expansion of TILs as an immunotherapeutic strategy, whereby TILs are harvested directly from tumor biopsies, expanded ex vivo, and then readministered to patients, referred to as adoptive cell therapy (ACT), with the main goal being to restore and enhance TIL anti-tumoral responses and the direct elimination of tumor cells
[79][80][81][145,146,147],
Tumor-derived EVs are known to promote tumor progression through their direct modulation and suppression of the host immune response and chemoresistance and peripheral tolerance in cancer patients
[82][83][148,149]. The cargo carried in tumor-derived exosome-like vesicles has been shown to include immunosuppressive molecules which influence the development, progression, and anti-tumor activity of immune cells either directly or indirectly
[84][150]. Nakazawa et al., 2021, demonstrated that tumor-derived EVs containing CD300a are taken up by dendritic cells resulting in their inhibited secretion of interferon-β (IFN-β) leading to enhanced tumor immunity via the decreased activation of regulatory T cells
[85][151]. Tumor-derived EV-dependent modulation of TIL activity occurs through the inhibition of proliferation and signaling activity of CD8+ T cells resulting in their apoptotic cell death
[86][152]. Contrarily, dendritic cell-derived EVs promote the proliferation of T cells within the TME
[87][153]. Cancer-derived CD8+ T cells from patients with head and neck cancer when co-cultured with tumor-derived EVs has been shown to induce the loss of CD27 expression in CD8+ T-cell and thus resulting in their change from the anti-tumor phenotype towards a more potent tumor suppressor phenotype
[88][89][90][154,155,156]. Additionally, several studies have also suggested that tumor-derived EVs expressing the transmembrane protein FasL isolated from the plasma of oral cancer patients have the ability to induce apoptosis of CD8+ T cells
[91][92][157,158]. Since regulatory T cells (Tregs) are critical for immune system suppression and the infiltration of Tregs into the TME, and their elevated presence in circulation is a strong prognostic marker in cancer
[93][159], several studies have demonstrated that tumor-derived EVs promote the expansion of CD4/CD25/FOXP3 triple-positive Tregs and apoptotic induction of TILs
[94][160]. Based on the immunoregulatory effects of EVs, which include the modulation of antigen presentation and immune activation and surveillance
[95][96][97][161,162,163], multiple studies have investigated the ability of exosome-like vesicles to participate in tumor regression, demonstrating that immune cell-derived EVs display potent cytotoxic effects in hepatocellular carcinoma when administered as a cell-free anti-tumor vaccine
[98][99][164,165].
Collectively, when considering the multifaceted and intricate role that EVs play in cell-to-cell communication between tumor cells, CAFs, TAMs, TECs, and TILs within the TME, it is no wonder that extensive research has been conducted to understand the mechanisms underlying EVs in tumor promotion and metastasis. Additionally, their enhanced secretion from tumors and the altered content of tumor-derived EVs when compared to EVs secreted from normal cells offer the potential to not only diagnose and monitor cancer progression, but also the generation of bioengineering EVs which interrupt the communication between tumor cells with the surrounding TME, thereby preventing EMT initiation, is an exciting and promising avenue in anti-metastatic therapy in cancer.