Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Razvan Adrian Covache-Busuioc and Version 2 by Lindsay Dong.
Aquaporins (AQPs), integral membrane proteins facilitating selective water and solute transport across cell membranes, have been the focus of extensive research over the past few decades. Particularly noteworthy is their role in maintaining cellular homeostasis and fluid balance in neural compartments, as dysregulated AQP expression is implicated in various degenerative and acute brain pathologies.
- aquaporins
- central nervous system
- fluid homeostasis
- glymphatic system
- neurodegenerative diseases
1. Introduction
1.1. Definition and General Characteristics of Aquaporins
Aquaporins represent a ubiquitous family of integral membrane proteins that are disseminated extensively across both animal and plant kingdoms. Structurally, these proteins are characterized by a core architecture comprising six alpha-helical transmembrane domains and two additional, shorter helical elements. These components delineate cytoplasmic and extracellular vestibules, which are connected via a constricted aqueous pore (Figure 1). Notably, conserved sequence motifs, such as the asparagine-proline-alanine (NPA) motifs, are frequently observed within these shorter helical regions. Functionally, aquaporin monomers autonomously integrate into the lipid bilayer, subsequently aggregating as tetrameric complexes. Certain variants, exemplified by mammalian Aquaporin 4 (AQP4), have the capability to further organize into higher-order structures, known as orthogonal arrays of particles, within the cell membrane [1][2][1,2].
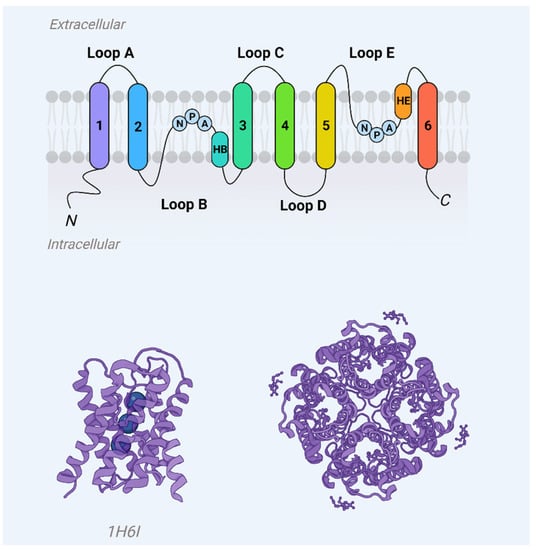
Figure 1. Aquaporin monomer membrane topography (upper panel) and crystal structure (AQP1, PDB 1j4n) showcase helices H1–H8 and water molecules (blue spheres) in the aqueous pore.
Additionally, a specialized subgroup of aquaporins, termed aquaglyceroporins, exhibits the capacity to transport glycerol molecules. The structural configuration of aquaglyceroporins is distinguished by a marginally larger pore diameter and a lining enriched with hydrophobic amino acid residues, in contrast to the hydrophilic nature of the pore in water-selective aquaporins. Beyond the primary functions of transporting water and, in some cases, glycerol, aquaporins are postulated to facilitate the passage of various gases (CO2, NH3, NO, O2) and small solutes such as hydrogen peroxide and arsenite. Some aquaporins have even been implicated in ion transport (K+, Cl−), although these claims remain a subject of ongoing debate [3][5].
Moreover, a range of non-transport-centric functions have been ascribed to some aquaporins, encompassing roles in cellular adhesion, membrane polarization, and the modulatory regulation of protein interactions, such as those involving ion channels [3][5].
1.2. The Overarching Significance of Aquaporins in Cellular Physiology, with an Emphasis on Their Role in the Central Nervous System
The expression profiles of Aquaporins (AQPs), particularly AQP4 and AQP1, have garnered significant research attention within the context of the central nervous system and sensory organs, as compared to the peripheral and enteric nervous systems [4][6]. AQP4, the predominant water channel in the brain, spinal cord, and optic nerve, is largely expressed in the astrocyte cell plasma membrane but is distinctly localized to specialized regions such as astrocyte foot processes [5][6][7][7,8,9]. Such polarized expression patterns are hypothesized to be mediated by intracellular associations between AQP4 and α-syntrophin or through extracellular interactions with agrin. Both α-syntrophin and agrin also exhibit polarized expression in astrocyte foot processes [8][10]. Additionally, the formation of orthogonal arrays of particles (OAPs) by AQP4 may facilitate its polarized localization, as it requires only a single anchoring link for stabilization, as opposed to individual AQP4 tetramers [8][10]. In the CNS, AQP4 is predominantly localized to the subpial astrocyte processes forming the glial-limiting membrane, the perivascular astrocyte endfeet, and the basolateral membrane of ependymal and subependymal astrocyte processes [9][10][11,12]. Such strategic localization suggests that AQP4 may play a critical role in regulating water flux across the CNS-water compartment interfaces. In specific brain regions lacking a blood–brain barrier, such as the circumventricular organs, and in hippocampal regions such as CA1 and the dentate gyrus, AQP4 manifests a more diffused expression profile [11][13]. This dispersed distribution may facilitate rapid water flux essential for maintaining potassium (K+) homeostasis during neuronal activity.23. Distribution and Molecular Classification of Aquaporins
2.1. Overview of Various Aquaporin Isoforms Present in Mammalian Systems
3.1. Overview of Various Aquaporin Isoforms Present in Mammalian Systems
In mammals, the aquaporin (AQP) family is generally made up of 12 to 15 isoforms grouped into 13 subfamilies (AQP0–12). However, humans stand out by having 18 paralogs due to tandem duplications, including four extra AQP7 pseudogenes and a second copy of AQP12. Additionally, more ancient mammalian lineages, such as Metatheria and Prototheria, have been shown to possess further classes such as AQP13–14 [12][37]. Animal AQPs are typically categorized into four main groups:-
Classical or Orthodox AQPs (AQP0, 1, 2, 4, 5, 6, 14), which are mainly focused on water transport.
-
Aqua-ammoniaporins (AQP8), which are sometimes counted among the orthodox AQPs.
-
Aquaglyceroporins (AQP3, 7, 9, 10, 13), which can transport glycerol in addition to water.
-
Plasma Membrane Intrinsic Proteins (PIPs);
-
Tonoplast Intrinsic Proteins (TIPs);
-
NOD26-Like Intrinsic Proteins (NIPs);
- Small Basic Intrinsic Proteins (SIPs);
- Unknown Intrinsic Proteins (XIPs), which are absent in monocots and Brassicaceae;
2.2. The Molecular Underpinnings of Their Selective Permeability and Functionality
3.2. The Molecular Underpinnings of Their Selective Permeability and Functionality
Water transport assays to study Aquaporins have been conducted using a variety of model systems across species, including bacteria, yeast, and mammalian cells [23][24][25][50,51,52]. This diversity in experimental setups reflects the widespread distribution of AQPs in nature. Specifically, cellular components such as intracellular and plasma membrane vesicles, particularly from animal tissues such as the kidney and intestinal epithelia, have been essential for evaluating AQP activity [26][53]. One of the prominent methods for characterizing new AQP isoforms involves the use of Xenopus laevis oocytes, owing to their inherently low water permeability [27][32]. This heterologous expression system allows for functional studies of AQPs from various organisms. Yeast cells lacking endogenous AQPs also serve as an effective model system for this purpose. They have been used to develop a high-throughput assay to identify functionally relevant AQP mutants that can withstand freeze-thaw cycles.2.3. Permeability Assays
3.3. Permeability Assays
The quantification of water or solute permeability across biological membranes serves as an indirect method to assess both the expression and functional status of Aquaporins. In general, the analytical focus for AQP activity and water permeability revolves around tracking alterations in cell or vesicle volume, which result from osmotically or pressure-driven water fluxes [28][56]. For solute permeability estimations, it is essential to account for both solute and water fluxes when examining volume changes, as these fluxes are instigated by their respective gradients. Both water and solute fluxes are directly correlated to their inducing forces, represented by osmotic permeability (Pf) and solute permeability (Ps) coefficients, respectively. The kinetics of volume changes depend on the partitioning of water or solute between the aqueous pathway through the channel and diffusion across the lipid bilayer. Furthermore, analyzing permeability as a function of temperature enables the calculation of the activation energy (Ea) required for transport, a crucial metric for gauging AQP functionality. It is generally observed that water or solute fluxes via hydrophilic channel pores necessitate lower activation energy compared to fluxes traversing a hydrophobic lipid bilayer. Analytical methodologies for assessing permeability commonly employ optical properties that are volume-dependent, such as light transmission, absorbance, scattering, and fluorescence [29][30][31][58,59,60]. These strategies are equally applicable to evaluating transport kinetics in both cellular models and proteoliposomes. The amalgamation of diverse biological models (cells, vesicles, and proteoliposomes) with optical detection systems offers a comprehensive toolkit for the investigation of AQP functions.2.4. Epithelial Assays
3.4. Epithelial Assays
Water permeability can be assessed in native epithelial tissues, such as those from the intestinal wall or kidney tubules, as well as in cultured epithelial cell monolayers positioned on permeable supports within Ussing chambers [32][61]. In these experiments, the apical and basolateral membranes of polarized cells are exposed to distinct compartments. The introduction of a membrane-impermeable solute such as sucrose or mannitol to one of these compartments instigates a transepithelial water flux. This flux is then quantified either by observing the fluid level change in a capillary tube linked to the opposite compartment or through the use of a fluorescent dye. The dye is added to the hyperosmotic compartment, and the rate of fluorescence alteration due to dye dilution serves as a measure of overall transepithelial osmotic water permeability [33][62].2.5. Osmotic Swelling Assays
3.5. Osmotic Swelling Assays
The functionality of AQP-mediated water transport can be probed using X. laevis oocytes in an osmotic swelling assay [28][56]. Oocytes that are microinjected with AQP mRNA are exposed to hypo-osmotic conditions, and the kinetics of cellular swelling are monitored using video microscopy. To investigate solute permeability, an inwardly directed chemical gradient is established, prompting a solute influx followed by an influx of water, culminating in oocyte swelling [34][63]. The low intrinsic water permeability and negligible permeability for glycerol and other solutes in oocytes make this system particularly well-suited for AQP studies.34. Functional Dynamics of Aquaporins in the Brain
3.1. Detailed Exploration of AQP4 and AQP1, Emphasizing Their Role in Maintaining Fluid Equilibrium across Neural Compartments
4.1. Detailed Exploration of AQP4 and AQP1, Emphasizing Their Role in Maintaining Fluid Equilibrium across Neural Compartments
Aquaporin 1, localized in the choroid plexus epithelial cells, and Aquaporin 4, found in ependymal cells as well as glial limitants, are implicated in the regulation of cerebrospinal fluid homeostasis and production. The specific roles played by these individual water channels in these processes are currently under academic debate. Evidence suggests that both AQP1 and AQP4 contribute significantly to CSF production and that a concurrent mutation in both AQP1 and AQP4 genes disrupts CSF drainage and ventricular compliance. Data further underscore the role of AQP4 in extra-choroidal CSF formation, advocating for a critical and sustained balance in CSF production and absorption, facilitated by water flux between brain capillaries and interstitial fluid (ISF). Cerebrospinal fluid and interstitial fluid form integral parts of the cerebral extracellular milieu, existing in a dynamic equilibrium that bathes neurons and glial cells. This fluid balance, maintained through a tightly regulated exchange between CSF and ISF, is essential for ensuring stable brain volume as well as ionic and solute concentrations for optimal neural and glial function [35][36][37][66,67,68]. Traditionally, CSF, which is synthesized from plasma modification, has been understood to occupy the brain ventricles and subarachnoid spaces and serve primarily as a mechanical cushion for the central nervous system (CNS). However, recent advancements have challenged this “classic view”, proposing an updated paradigm wherein: (i) extra-choroidal CSF production occurs through fluid exchange between brain capillaries and ISF; (ii) there exists noteworthy paravascular CSF/ISF flow within the brain parenchyma; and (iii) the meningeal lymphatic system plays a significant role in CSF drainage [37][38][39][68,70,71]. Aberrations in CSF homeostasis are strongly correlated with the pathophysiology of various CNS conditions, including hydrocephalus, cerebral edema, and ischemia. Cerebral aquaporins, specifically AQP1 and AQP4, are posited to be crucial regulators of cerebrospinal fluid (CSF) and interstitial fluid (ISF) homeostasis. Given their expression profiles—AQP1 localized solely in the choroid plexus epithelial cells and AQP4 in ependymal cells as well as glial limitants—it has been simplistically theorized that AQP1 is primarily involved in CSF production while AQP4 facilitates CSF/ISF exchange and absorption [40][74]. Consistent with traditional theories of CSF circulation, AQP1-deficient mice exhibited reduced CSF production and intraventricular pressure, underscoring the importance of AQP1 in CSF genesis [41][75].3.2. Deep Dive into the Interplay between Aquaporins, Cerebrospinal Fluid, and the Intricacies of Brain Fluid Homeostasis
4.2. Deep Dive into the Interplay between Aquaporins, Cerebrospinal Fluid, and the Intricacies of Brain Fluid Homeostasis
Aquaporin 1 (AQP1) and Aquaporin 4 (AQP4) serve as key water channel proteins in the central nervous system, each exhibiting unique but complementary expression patterns within cerebral tissues. Both are postulated to be central regulators of cerebral fluid homeostasis under both physiological and pathological conditions. Specifically, AQP1 is localized solely to the apical membranes of choroid plexus epithelial cells, while AQP4 is predominantly expressed in astrocytes and ependymal cells [4][6]. Traditionally, the choroid plexus has been viewed as the primary site for cerebrospinal fluid (CSF) production, facilitated by AQP1, with the fluid then flowing unidirectionally into the subarachnoid space for eventual reabsorption via arachnoid granulations. However, emerging evidence underscores the significance of extra-choroidal CSF formation, whereby CSF is generated through water filtration from brain parenchymal capillaries, facilitated by AQP4 [42][76].45. The Glymphatic System: An Essential Framework for Brain Health
4.1. Comprehensive Breakdown of the Glymphatic System’s Architecture and Its Functional Significance
5.1. Comprehensive Breakdown of the Glymphatic System’s Architecture and Its Functional Significance
The glymphatic system, a groundbreaking discovery in neuroscience, serves as a macroscopic waste clearance pathway that employs a specialized network of perivascular channels created by astroglial cells. This system is essential for the efficient removal of soluble proteins and metabolites from the central nervous system. Beyond its role in waste clearance, the glymphatic system may also assist in the distribution of vital substances such as glucose, lipids, amino acids, and neurotransmitters across the brain. Notably, the system is primarily active during sleep, suggesting that the biological necessity for sleep may stem from the brain’s need to eliminate potentially harmful waste products, including β-amyloid [43][87]. This influx of CSF into the brain parenchyma triggers convective fluxes of ISF within the tissue, leading it toward perivenous spaces adjacent to large, deep veins. Eventually, the ISF is gathered in these perivenous spaces and drains towards the cervical lymphatic system [44][45][89,90]. The system of convective fluid fluxes, characterized by its rapid exchange between CSF and ISF, has been dubbed the glymphatic system. This name reflects its functional similarities to the peripheral lymphatic system and emphasizes the critical role of glial AQP4 channels in facilitating fluid transport. Subsequent analyses revealed that this directed movement of CSF via periarterial pathways into the brain parenchyma assists in clearing out interstitial solutes, directing them towards perivenous drainage channels. This finding has significant implications for neurodegenerative diseases, such as Alzheimer’s, which are characterized by protein accumulations, including amyloid plaques and tau tangles [46][47][91,92]. Moreover, imaging studies in mice lacking Aquaporin 4 (AQP4) showed significant disruptions in the glymphatic system. Specifically, there was approximately a 65% reduction in the flux of CSF through the brain tissue when compared to wild-type control mice. Additionally, the clearance rate of intrastriatally-injected radiolabeled β-amyloid was reduced by 55% [39][71].4.2. AQP4-Centric Discussion on the Glymphatic Pathway, Detailing How Aquaporin Malfunctions Might Hinder the System
5.2. AQP4-Centric Discussion on the Glymphatic Pathway, Detailing How Aquaporin Malfunctions Might Hinder the System
Convective CSF Fluxes in Aging and Pathology
Glymphatic activity decreases sharply during aging-Recent research has shown a striking decrease in glymphatic function in older mice compared to their younger counterparts, with an estimated 80–90% reduction in activity [48][93]. This decline encompasses both the influx of CSF tracers into the brain and the clearance of radiolabeled β-amyloid and inulin. It has been suggested that reactive gliosis, characterized by the hypertrophy of GFAP+ astrocyte processes, may be a contributing factor to this age-related decline in glymphatic function. However, the exact mechanisms linking changes in GFAP expression to diminished glymphatic activity remain unclear [49][94]. In younger animals, Aquaporin 4 is localized to the astrocytic endfeet and plays a pivotal role in facilitating the exchange of cerebrospinal and interstitial fluid along periarterial influx pathways (Figure 2), as well as aiding in the clearance of interstitial solutes through perivascular drainage paths. Previous research has shown that the genetic deletion of AQP4 leads to a roughly 65% impairment in CSF-ISF exchange and a 55% reduction in the clearance of β-amyloid [50][95]. In aging brains, there is a partial loss of this vascular polarization of astrocytic AQP4 (Figure 2). Specifically, AQP4 is no longer solely confined to the astrocytic endfeet but is also found in the parenchymal processes of astrocytes [51][96].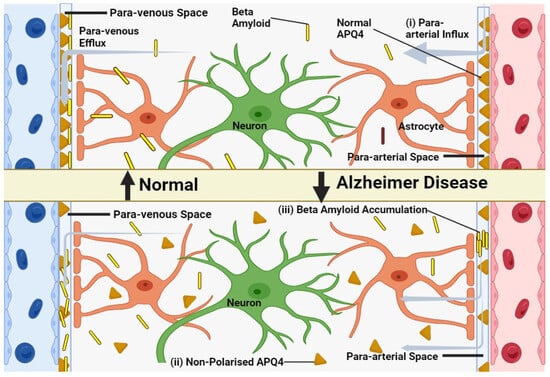
Figure 2. Model of glymphatic function in young, old and in Alzheimer’s disease. (i) CSF efficiently clears brain solutes in young, healthy individuals via periarterial pathways. (ii) Aging disrupts glymphatic function, possibly due to reactive astrocytes and AQP4 de-polarization. (iii) In Alzheimer’s, β-amyloid accumulates in perivascular spaces, potentially due to glymphatic impairment, further hindering waste clearance.
4.3. Correlation between Compromised Glymphatic Functionality and Neurodegenerative Disorders
5.3. Correlation between Compromised Glymphatic Functionality and Neurodegenerative Disorders
Traumatic brain injuries (TBIs), commonly observed in military personnel and athletes, are associated with an elevated risk of premature dementia and Alzheimer’s disease [52][99]. Numerous studies have indicated that both recurring traumatic incidents, as well as single events of moderate to severe head trauma, can result in ongoing neurodegeneration. The mechanism by which only certain individuals develop chronic traumatic encephalopathy after experiencing a similar degree of initial brain injury remains elusive [53][100]. TBIs result in the release of β-amyloid peptide and C-tau, a proteolytic derivative of MAP-tau, which is a prominent intracellular microtubule protein in axons. Notably, C-tau, due to its vast release correlating with TBI severity, serves as a brain injury biomarker [54][101]. A prevailing theory posits that significant surges in interstitial tau can lead to its cellular intake, initiating fibrillary aggregates. These aggregates attract more tau, fostering the formation of neurofibrillary tangles and facilitating a prion-like progression of the disease. Moreover, TBIs are connected to the development of extensive astroglial scars and the sustained activation of inherent neuroinflammation.
56. Implications of Aquaporins in Degenerative and Acute Brain Pathologies
5.1. Profiling Each Degenerative Disease (iNPH, PD, AD) and Its Associated Aquaporin Dysregulations
6.1. Profiling Each Degenerative Disease (iNPH, PD, AD) and Its Associated Aquaporin Dysregulations
56.1.1. Parkinson’s Disease
Neurodegenerative diseases impact millions globally, constituting a diverse set of disorders that specifically target certain regions of the Central Nervous System (CNS). These diseases typically result in a sustained decline in cognitive or motor functions, contingent upon the distinct type of neuronal cells undergoing selective degeneration [55][103]. Predominantly, these pathological states are correlated with aging. Indeed, as life expectancy has risen over recent decades, the prevalence of these age-related disorders has correspondingly increased [56][104]. Neuronal harm and oxidative stress, which are fundamental events in the onset of these conditions, precipitate an upsurge in the production of pro-apoptotic and pro-inflammatory cytokines.56.1.2. Dopamine Regulation of AQP4 Expression
In the realm of neurodegenerative diseases, a pivotal role is played by neural stem cells. Throughout adulthood, these cells have the capability to proliferate and differentiate into either new neurons or glial cells [57][106]. Intriguingly, studies have highlighted that adult neural stem cells display glial-associated properties in both in vivo and in vitro environments. As a testament to this, these stem cells express GFAP, a protein considered a hallmark for fully differentiated astrocytes [58][107]. Notably, alterations in the count of GFAP-expressing cells have been implicated in neurodegenerative disorders, including Parkinson’s disease [59][108]. Dopamine (DA), a central neurotransmitter, has been found to invigorate the proliferation of progenitor cells. This effect is observed not just in the striatum but also in the subventricular zone of mature brains. Building on this, a recent investigation led by Kueppers et al. postulates that DA orchestrates the proliferation of striatal astrocytes in culture through the mediation of Aquaporin-4 [60][109]. Their results delineate a scenario wherein DA prompts a reduction in AQP4 expression in striatal glial cells when studied in vitro.56.1.3. Mitochondrial AQP9 in PD Brains
Within the scope of neurodegenerative disorders, a potentially significant yet conjectural association has been proposed between Aquaporin-9 (AQP9) and Parkinson’s disease [61][113]. Within the cerebral environment, AQP9—a channel facilitating the movement of water and certain solutes—is manifested in astrocytes, brain stem catecholaminergic neurons, as well as specific subgroups of midbrain dopaminergic and hypothalamic neurons. An intriguing observation is the pronounced presence of AQP9 within the mitochondrial inner membranes, hinting at its potential role in supporting neuronal metabolism. Especially compelling is the notion that aberrations in mitochondrial AQP9 within dopaminergic neurons might be linked to the heightened susceptibility of these neurons to PD [61][113].56.1.4. Idiopathic Normal Pressure Hydrocephalus (iNPH)
Idiopathic normal pressure hydrocephalus (iNPH) is recognized as a distinct form of dementia, wherein intervention through cerebrospinal fluid diversion can yield favorable outcomes. Pioneering studies leveraging magnetic resonance imaging with CSF tracers have highlighted compromised CSF tracer clearance from specific cerebral regions, notably the entorhinal cortex, in iNPH patients. This compromised clearance mechanism, especially for waste solutes such as soluble amyloid-β, might be central to the neurodegenerative processes and cognitive decline characterizing iNPH. The primary objective of the study at hand was to investigate potential alterations in the subcellular localization of aquaporin-4 water channels in relation to iNPH. Notably, AQP4 is known to play a pivotal role in modulating CSF flow and facilitating the glymphatic removal of brain metabolites. To accomplish this, cortical brain biopsy samples from 30 iNPH patients and 12 control subjects were scrutinized using AQP4 immunogold cytochemistry. Utilizing electron microscopy, the study unveiled a markedly diminished presence of AQP4 water channels within the astrocytic endfoot membranes juxtaposed to cortical microvessels in iNPH patients, relative to the control group. Further analysis divulged a statistically significant association between AQP4 densities oriented perivascularly and those directed towards the parenchyma.5.2. Insights into Aquaporin Behavior during Acute Cerebral Events, Such as Stroke or Traumatic Injuries
6.2. Insights into Aquaporin Behavior during Acute Cerebral Events, Such as Stroke or Traumatic Injuries
Aquaporins, water channel proteins found in the brain, have attracted substantial research attention given their potential implications in both physiological and pathological contexts [62][117]. Among these, AQP4 has been the focal point of numerous studies, especially in relation to various brain conditions, spanning acute afflictions such as stroke and traumatic brain injury to chronic autoimmune neurodegenerative ailments. As of now, there are no targeted therapeutic interventions specifically designed to modulate the water transport activity of these channels. However, accumulating experimental data underscores the pivotal nature of AQPs, suggesting their profound relevance in future research endeavors A predominant characteristic of many brain diseases, ranging from stroke, traumatic brain injuries, and brain tumors to inflammation, is edema. This phenomenon denotes the accumulation of water due to imbalances in brain osmotic homeostasis. A primary ramification of edema is brain swelling, which can exacerbate secondary complications, such as compromised brain perfusion. Despite its long-standing recognition in both clinical and pre-clinical realms, the molecular and cellular intricacies underpinning edema genesis and resolution remain relatively elusive. Furthermore, current therapeutic interventions fall short of effectively curbing edema onset or progression in various brain conditions.56.2.1. Edema Build-Up Phase: Anoxic, Ionic and Vasogenic Edema
Cerebral edema, for the past four decades, has been predominantly categorized into two principal types: cytotoxic and vasogenic [63][119]. Conventionally, cytotoxic edema denotes the accumulation of intracellular water without any disruption of the blood–brain barrier (BBB). In contrast, vasogenic edema emerges post-BBB disruption, instigating protein diffusion from the bloodstream into the tissue, subsequently leading to water buildup in the extracellular matrix. This longstanding bifurcation, however, is now considered an oversimplification, particularly in light of recent insights into the molecular shifts during edema onset and the evolving understanding of BBB properties. Anoxic edema is typified by the immediate swelling of astrocytes and neuronal dendrites ensuing moments after a deprivation of oxygen and glucose, particularly in cerebrovascular ailments. This deficiency in essential nutrients triggers significant perturbations in cellular ionic gradients due to the non-operational energy-dependent co-transporters. Consequently, this facilitates an extensive ion influx into cells, a manifestation of which is a gradual surge in the extracellular K+ levels, leading to subsequent water entry and resultant swelling initially in astrocytes and later in neuronal dendrites [64][121].56.2.2. Contribution of AQPs in Edema Formation and Resolution
Aquaporins, specifically AQP1, 4, and 9, have demonstrated alterations in their expression levels in various brain disorders, as seen in both rodent models and human specimens [65][125]. Of these, AQP4 has been the primary focus of many investigations, as its expression patterns appear to mirror the progression of edema in numerous neurological conditions [66][126]. A significant advancement in the exploration of AQP4’s role in edema came with the creation of AQP4 knockout mice (AQP4−/−) by Dr. Verkman’s team [67][127].56.2.3. AQP4 and Edema Build-Up
The role of Aquaporin-4 in cerebral edema is complex, exhibiting a spectrum of expression profiles across various pathological conditions including traumatic brain injury, ischemic stroke, and subarachnoid hemorrhage. These conditions each display unique fluctuations in AQP4 expression levels [66][68][69][124,126,129]. Specifically, in ischemic models involving transient middle cerebral artery occlusion, AQP4 is acutely up-regulated in the astrocyte endfeet adjoining blood vessels, reaching peak levels approximately 1 h post-stroke onset. This elevation in AQP4 expression is spatially and temporally correlated with the extent of cerebral edema and is most prominent in the peri-infarct region as well as the prospective lesion site [66][70][123,126].56.2.4. Edema Resolution in Acute Brain Disease: Role of AQP in Water Clearance
The hypothesis that Aquaporin-4 has a dual role in cerebral edema—being deleterious during edema formation while beneficial during its resolution—has gained empirical support despite the absence of conclusive evidence. Initial evidence for this comes from experiments using AQP4 knockout mice (AQP4−/−), which revealed that intracranial pressure increased significantly when a saline solution was infused into the brain parenchyma compared to wild-type mice [6][8]. Specifically, AQP4 expression typically escalates 48 h post-insult in models of stroke, TBI, and neuroinflammatory lesions. This augmented expression is frequently localized to the astrocyte endfeet adjacent to blood vessels, as well as the astrocyte processes and glia limitans [66][69][126,129]. Such spatial distribution of heightened AQP4 expression suggests its potential role in facilitating the clearance of edematous fluid via the subarachnoid space. The role of other aquaporin family members, such as Aquaporin-9, in the resolution of cerebral edema warrants exploration. While AQP9 is up-regulated in reactive astrocytes seven days post-ischemic injury along the infarct border, its expression pattern does not show a strong correlation with the extent of cerebral swelling, in contrast to AQP4 [71][136].56.2.5. Chronic Changes of Brain AQP: Relation with Water Homeostasis Dysfunction?
Aquaporins, traditionally implicated in water homeostasis both in physiological and pathological contexts, have been increasingly associated with a broader range of cellular functions, including cell migration and gas diffusion [72][140]. Specific isoforms, such as AQP1, AQP4, and AQP9, manifest elevated expression in both brain tumors and peritumoral tissues. The augmented presence of these AQPs in astrocytes within peritumoral regions may be related to edema formation, potentially due to altered tissue homeostasis and elevated metabolic rates. Additionally, these AQPs may facilitate gas diffusion, playing a role in the clearance of excess CO2 and the diffusion of O2. In rodent models of spinal cord injury (SCI), an upregulation of AQP1 is observed in astrocytes as well as neurons. AQP1 expression has also been documented in neuronal processes in the dorsolateral septum at various time points following juvenile traumatic brain injury (jTBI) [69][129]. While astrocytic expression of AQP1 may be associated with cerebrospinal fluid secretion during cyst formation, its co-localization with growth-associated protein-43 (GAP-43) in neurons suggests a role in neuroplasticity and repair after injury.5.3. The Cascading Effects of Altered AQP Expression in Autoimmune Conditions, with a Focus on NMO
6.3. The Cascading Effects of Altered AQP Expression in Autoimmune Conditions, with a Focus on NMO
Aquaporins are not just pivotal in regulating water homeostasis; they also have roles in the immune system, specifically within both the innate and adaptive arms. In human blood leukocytes, AQP1 and AQP9 are expressed and show upregulation upon stimulation with lipopolysaccharide (LPS) either intravenously or in vitro [73][144]. AQP9 expression is also elevated in activated polymorphonuclear leukocytes in patients suffering from systemic inflammatory response syndrome (SIRS) and infective endocarditis [74][145]. B and T lymphocytes, key players in adaptive immunity, have been found to express AQP1, AQP3, and AQP5. Similarly, immature dendritic cells (DCs), which are integral to the innate immune system, express AQP3 and AQP5. In these immune cells, AQP expression is often correlated with their activation and proliferation. Notably, AQP9 is the most highly expressed isoform in DCs and shows further upregulation upon LPS stimulation. Human primary blood-derived macrophages and neutrophils, critical components of the innate immune system, display high levels of AQP9, which also sees upregulation at both transcript and protein levels when stimulated with LPS [75][147]. AQP3, another isoform, is also sensitive to LPS stimulation in monocytic THP-1 cells, a model often used to study inflammation. The inhibition or silencing of AQP3 in these cells leads to partial blockage of LPS priming and a reduction in the production of key inflammatory cytokines such as interleukin-6 (IL-6), pro-IL-1β, and tumor necrosis factor-alpha (TNF-α). Neuromyelitis optica spectrum disorders (NMOSD) are a range of inflammatory demyelinating diseases (IDDs) that primarily affect the optic nerves and spinal cord but can also extend to the brain and, in rare instances, muscles. Brain lesions in NMOSD often localize to areas with high AQP4 expression, such as the circumventricular organs (responsible for intractable nausea and vomiting) and the diencephalon (linked to sleep disorders, endocrine imbalances, and the syndrome of inappropriate antidiuresis). Up to 10% of NMOSD patients even fulfill the Barkoff criteria for multiple sclerosis when evaluated through MRI [76][153]. One of the hallmarks of NMOSD is the presence of autoantibodies against AQP4, known as AQP4-IgG or NMO-IgG, detected in 60–90% of NMO patients [77][78][154,155]. AQP4 is not only found on the astrocytes in the central nervous system (CNS) but also in skeletal muscle and various epithelial cells such as those in the kidney, stomach, and exocrine glands. AQP4-IgG was initially perceived as a mere marker of the disease, possibly related to astrocyte damage. However, mounting evidence now suggests that AQP4-IgG plays a pathogenic role in NMO. When AQP4-IgG binds to AQP4 on astrocytes, this initiates a cascade of immunological responses, primarily through complement-dependent cytotoxicity. This leads to the invasion of leukocytes into the CNS, the release of cytokines, and the disruption of the blood–brain barrier. These series of events are thought to culminate in the death of oligodendrocytes (cells responsible for myelination), resulting in myelin loss and, ultimately, neuronal death. This cascade explains the neurological deficits observed in patients with NMO. Given the rapidly evolving knowledge on the immunobiology of AQP4 autoimmunity, there is a growing need for ongoing revisions in the diagnostic criteria for NMOSD [76][153]. As our understanding broadens, the role of highly specific assays that can detect pathogenic AQP4-IgG targeting the extracellular domains of AQP4 becomes increasingly crucial. The size of AQP4-IgG (autoantibodies against AQP4) presents a steric challenge when it comes to binding with AQP4, which is a tetramer consisting of four separate monomers and, by extension, four distinct water pores. This size mismatch suggests that it would be unlikely for a single AQP4 tetramer to bind with more than one AQP4-IgG molecule. Therefore, significant inhibition of water permeability by AQP4-IgG appears to be theoretically implausible.67. Aquaporins at the Intersection of Oncology and Neurology
6.1. Unraveling the Possible Links between Aquaporin-Mediated Processes and Brain Tumorigenesis
7.1. Unraveling the Possible Links between Aquaporin-Mediated Processes and Brain Tumorigenesis
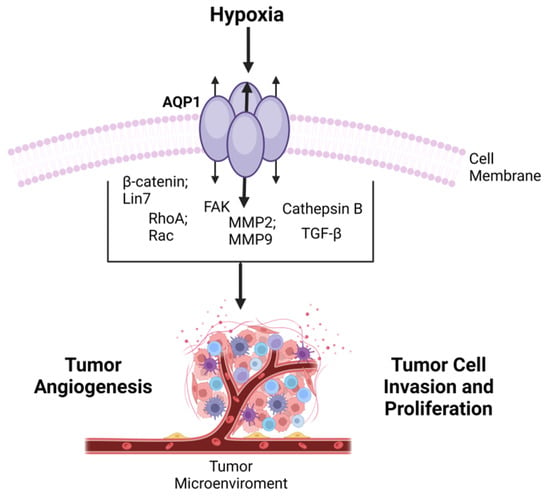
AQP1, primarily known for its role in water transport, has additional functions that are intriguing both scientifically and medically. One of these is its capability to act as a cyclic nucleotide-gated cation channel activated mainly by cGMP and, to a lesser extent, by cAMP [79][160]. Research by Yu and colleagues suggests that the interaction of cGMP with an arginine-rich cytoplasmic Loop D in AQP1 leads to a conformational change that may mediate the gating of its central ion channel [80][81][161,162]. The link between AQP1 and cancer progression has garnered significant attention, leading to numerous reviews on the subject. These reviews often focus on the potential of AQP inhibitors as therapeutic agents in cancer treatment [82][83][166,167]. Given the multifaceted roles of AQP1, from water transport to ion channel gating and its association with various types of cancer, the protein appears to be a critical player in both physiology and pathophysiology, including tumorigenesis. As such, understanding its function and regulation could offer valuable insights into the development of novel therapeutic approaches for a range of diseases, including cancer (Figure 34).Figure 34. Overview of AQP1 in Cancer Progression-tumor development via transited molecules-emphasizing its involvement in cell migration, invasion, and angiogenesis, and its potential as a prognostic factor in various cancers.
6.2. Implications of Aquaporins in Neoplastic Cell Migration, Invasiveness, and Angiogenesis
7.2. Implications of Aquaporins in Neoplastic Cell Migration, Invasiveness, and Angiogenesis
67.2.1. AQP1-Modulated Tumor Cell Migration and Invasion
The role of AQP1 in cancer progression extends beyond its well-known function in water transport, particularly implicating it in the critical processes of tumor cell migration and invasion. Hu and Verkman found that AQP1 accelerates the migration of specific mouse melanoma and breast cancer cell lines in vitro. Notably, they observed polarized AQP1 expression at the leading edge of migrating cells. In vivo studies further revealed that AQP1 promoted cancer cell extravasation and lung metastases [84][168].
One proposed mechanism by which AQP1 facilitates tumor cell migration involves osmotic water flow across the plasma membrane. This flow is thought to be induced by an osmotic gradient created by actin depolymerization and active solute influx at the cell’s leading edge [85][170]. Water influx via AQP1 then increases hydrostatic pressure, leading to local expansion of the plasma membrane and actin re-polymerization to stabilize cell membrane protrusions [4][6].
An alternative explanation focuses on AQP1’s role in changing the cell shape and volume as tumor cells navigate through confined spaces. The water flow facilitated by AQP1 helps generate the hydrostatic forces needed for this process. Actin polymerization and depolymerization as well as ionic fluxes across the membrane could support this osmotic water flow [68][124].
Adding a more nuanced layer to these mechanisms, Stroka and colleagues proposed an “Osmotic Engine Model”, where cell migration in confined spaces is driven not just by actin and myosin interactions but also through water permeation and ion transport mediated by AQPs and Na+/H+ pumps [86][172].
The implications of these findings are profound, as the acquisition of migratory and invasive capabilities is a key step in cancer metastasis, which accounts for the majority of cancer-related deaths. Understanding AQP1’s multifaceted role in these processes could thus offer crucial insights for the development of new therapeutic strategies targeting cancer metastasis.
These ion channels and transporters are implicated in crucial stages of tumor metastasis, such as loss of cell-to-cell contacts, invasion of the surrounding stroma, and entry into and exit from blood vessels (intra- and extra-vasation) [87][175]. Kourghi and colleagues further deepen this narrative by showing that certain AQP1 ion channel blockers, which do not impact AQP1’s water channel activity, effectively inhibited the migration of HT29 cancer cells. The degree of inhibition was directly related to the potency of the AQP1 ion channel blockage, suggesting that the ion channel properties of AQP1 alone might be sufficient for facilitating tumor cell migration in certain cases [88][176].